
ABSTRACT
MTORC1 is a well-known key regulator of macroautophagy/autophagy. However, the underlying regulatory mechanisms of MTORC1 activity remains elusive. We showed recently that SHOC2, a RAS activator, competes with MTOR for RPTOR (but not RICTOR) binding, leading to MTORC1 inactivation, autophagy induction and cell survival, whereas RPTOR competes with RAS for SHOC2 binding to inactivate RAS-MAPK and suppresses growth. Interestingly, SHOC2 is subjected to FBXW7 regulation. Upon growth stimulation, MAP2K1 phosphorylates SHOC2 on T507 to facilitate its binding with FBXW7B/FBXW7β for ubiquitination and degradation to terminate growth signaling, thus establishing a negative feedback loop. Human cancers with FBXW7 inactivation and SHOC2 overexpression would squeeze RPTOR from MTORC1, leading to MTORC1 inactivation and autophagy induction. Collectively, we propose a new mode of the FBXW7-SHOC2-RPTOR axis in control of MTORC1 activity that affects autophagy and cancer cell survival.
Autophagy, a cellular process mobilizing intracellular nutrient resources, plays an important role in cell survival during unfavorable growth conditions. MTOR is a key component that coordinately regulates the fine balance between growth and autophagy in response to cellular physiological conditions and environmental stresses such as deprivation of amino acids and glucose. Despite the significant progress in the study of autophagy, the mechanism by which MTOR activity is precisely regulated to control autophagy remains elusive. To this end, we conducted an immunoprecipitation assay and detected SHOC2 in both RPTOR and MTOR immunoprecipitates, while MTOR and RPTOR, but not RICTOR, were detected in SHOC2 immunoprecipitates [Citation1]. The results indicate that SHOC2 and RPTOR bind together under physiological conditions, and that SHOC2 via RPTOR binding interacts with MTORC1, but not MTORC2 due to lack of binding with RICTOR. We next determined whether the SHOC2-RPTOR binding would affect RPTOR-MTOR binding, and found that SHOC2 competes with MTOR for RPTOR binding through the middle and C-terminal regions, thus depleting RPTOR from the MTORC1 complex to inactivate MTORC1 and induce autophagy.
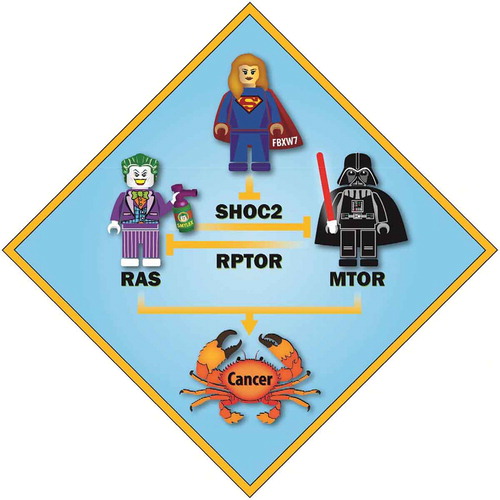
Figure 1. Negative cross-talk between RAS and MTORC1, controlled by FBXW7. RAS and MTORC1 are 2 oncogenic pathways and their activation leads to cancer development. In a recent study we showed that RAS (shown as the Joker) and MTORC1 (shown as Darth Vader) negatively cross-talk with each other, which is regulated by the tumor suppressive FBXW7 E3 ligase (shown as Superwoman). Specifically, SHOC2, a RAS activator, competes with MTOR for RPTOR binding to inactivate MTORC1. Similarly, RPTOR competes with RAS for SHOC2 binding to inactivate RAS. FBXW7 controls both RAS and MTORC1 pathways via promoting ubiquitination and degradation of SHOC2. Thus, the FBXW7-SHOC2-RPTOR axis precisely regulates proliferation and autophagy. (Image created by Steven Kronenberg.).
SHOC2/Sur8 was first identified in C. elegans as SOC-2, a scaffold for homologs of RAS and RAF to positively regulate the equivalent of the RAS-MAPK/ERK signaling pathway. SHOC2 is an evolutionarily conserved protein, composed of an unstructured N-terminal domain and a long stretch of leucine-rich repeats (LRR). The N-terminal domain binds to RAS and RAF to activate MAPK1/ERK2-MAPK3/ERK1. Given its role as a RAS and RAF activator, it is not surprised that SHOC2 is overexpressed in a number of human cancers. Interestingly, however, in human cancer cells with constitutively active RAS, SHOC2 still promotes anchorage-independent growth, clonal survival, and xenograft tumor growth in nude mice, whereas SHOC2 knockdown inhibits MAPK, demonstrating its oncogenic activity independent of RAS activation. Given its biological significance in regulation of growth (via RAS-MAPK) and autophagy (via RPTOR-MTORC1), it is important to understand how SHOC2 turnover is regulated and whether it is abnormally regulated in human cancer. Although HUWE1 E3 ligase was reported to ubiquitinate SHOC2, it is, however, not for the purpose of SHOC2 degradation, but for facilitating RAF ubiquitination and degradation.
To identify the E3 ubiquitin ligase that controls the turnover of SHOC2, we first determined whether SHOC2 is accumulated by treatment with MLN4924, a small molecule inhibitor of SCF E3 ligases. Indeed, MLN4924 causes a dose-dependent accumulation of SHOC2, suggesting involvement of SCF E3 ligase. Because SHOC2 is an oncogenic protein, we hypothesized that it is likely ubiquitinated and degraded by a tumor suppressive F-box protein, such as FBXW7. FBXW7, a haplo-insufficient tumor suppressor, is the substrate-recognizing subunit of SCF E3 ubiquitin ligase, which promotes ubiquitination and degradation of several oncoproteins, including MYC/c-Myc, MCL1, JUN/c-Jun, NOTCH1, CCNE/cyclin E and NFKB2/NFκB2/p100. FBXW7 interacts with a specific conserved phospho-degron sequence ((L)-X-pT/pS-P-(P)-X-pS/pT) on its substrates. A consensus binding motif search on SHOC2 identified 2 evolutionarily conserved sites at residues 240–243 (LITL) and 505–508 (LLTH), of which the second site was confirmed as being responsible for FBXW7 binding. Subsequent characterizations revealed that FBXW7 pulled down endogenous SHOC2 and shortened its half-life by promoting its ubiquitination and degradation. Thus, SHOC2 is a new oncogenic substrate of FBXW7.
Phosphorylation of a substrate at its F-box binding motif is prerequisite in most cases for FBXW7 binding for targeted ubiquitination. Because FBXW7 binds to and promotes ubiquitination of both wild-type SHOC2 and its T242A mutant, but not the T507A mutant, we reasoned that the residue Thr507, but not Thr242, is likely being phosphorylated prior to FBXW7 binding. To indentify the signal that could trigger SHOC2-Thr507 phosphorylation, we used computer software GSP3.0, which predicts MAP2K1/MEK1 as a kinase for Thr507 phosphorylation with a high probability. Following this lead, we confirmed that the MAPK signal, activated by growth factors, such as EGF and serum, indeed triggers SHOC2 Thr507 phosphorylation to facilitate FBXW7 binding and subsequent ubiquitination, which is blocked by MAPK inhibitor. These results indicate the MAPK signal is responsible for SHOC2 Thr507 phosphorylation.
What is the biological significance of SHOC2-triggered autophagy? We found that both SHOC2-stimulated cell growth and clonal survival are significantly reduced if autophagy is blocked (e.g., via ATG5 knockdown). Thus, SHOC2-induced autophagy appears to provide nutrients for cell proliferation, explaining a RAS-activation independent function of SHOC2. Moreover, the balance for overall cell growth and autophagy, mediated by the SHOC2-RPTOR-MTORC1 axis, has an upstream regulator, FBXW7, via targeted degradation of SHOC2. Thus, an accelerated growth is expected in human cancers, particularly with FBXW7 loss-of-function mutations and SHOC2 overexpression. The bioinformatics mining of human cancer databases at both the genomic and expression levels reveals missense and truncating mutations of SHOC2 in human lung cancer. Two mutants with potential alteration of surface structure were characterized, and both promote proliferation and survival of lung cancer cells, indicating that these mutants are not random, but have biological significance.
In summary, our study presents 3 new findings: First, SHOC2 is a new oncogenic substrate of FBXW7, providing an additional mechanism of FBXW7 action as a tumor suppressor, and also identifying the first E3 ligase that regulates SHOC2 turnover; second, a negative feedback-loop exists that terminates the MAPK signal by FBXW7-mediated SHOC2 degradation upon SHOC2 phosphorylation by MAP2K1; third, a negative cross-talk exists between the signals of SHOC2-RAS and RPTOR-MTORC1. SHOC2-mediated inactivation of MTORC1 triggers autophagy to provide nutrients for proliferation, whereas RPTOR-mediated inactivation of RAS-MAPK suppresses growth. shows how the FBXW7-SHOC2-RPTOR axis regulates the cross-talk of RAS-MTORC1 for cancer development.
Disclosure statement
No potential conflict of interest was reported by the authors.
Reference
- Xie CM, Tan M, Lin XT, et al. The FBXW7-SHOC2-raptor axis controls the cross-talks between the RAS-ERK and mTORC1 signaling pathways. Cell Rep. 2019 Mar 12;26(11):3037–3050.e4.