
ABSTRACT
Numerous studies have reported that inhibition of MTOR (mechanistic target of rapamycin kinase) clearly reduces Alzheimer disease neuropathological hallmarks in mouse models. This has resulted in calls for the use of the MTOR inhibitor rapamycin for the treatment of dementia in humans. Unfortunately, intervention with rapamycin in these mouse studies commenced before or early in the appearance of these pathological hallmarks. Later in Alzheimer disease, when dementia actually manifests, the brain’s lysosomal system is severely damaged and treatment with rapamycin is likely to exacerbate this damage. We reassess literature described by a recent perspective article calling for the use of MTOR inhibition in dementia and conclude that rapamycin could be useful, but only in people who are in the earliest stages of Alzheimer disease. We contend that our interpretation of preclinical data concerning use of rapamycin in Alzheimer disease models is necessary if we are to avoid another failed Alzheimer disease drug trial.
Abbreviations
AD: Alzheimer disease; APP: amyloid beta precursor protein; MAPT: microtubule associated protein tau; MTOR: mechanistic target of rapamycin kinase; MTORC1: mechanistic target of rapamycin kinase complex 1.
The recent article in Science Translational Medicine by Kaeberlein and Galvan [Citation1] explained the case for inhibition of mechanistic target of rapamycin kinase complex 1 (MTORC1) as a therapeutic intervention for Alzheimer disease (AD). They correctly pointed out that rapamycin – a highly-specific allosteric partial inhibitor of MTORC1 – consistently reduces amyloid plaque deposition and MAPT/tau (microtubule associated protein tau) aggregation in transgenic mouse models of AD. The authors then outline attitudes in the scientific community that have precluded the use of rapamycin in clinical trials for AD. Here, we discuss why the use of rapamycin in AD could indeed be beneficial but requires very careful consideration in the light of data from research on the lysosomal system.
Rapamycin pushes proteostasis towards catabolism, in part through augmentation of lysosomal-system function, a widely known phenomenon. The lysosomal system is the cell’s recycling center, which hydrolyses organelles and macromolecules to their constituent parts. These cargos are delivered via lysosomal pathways such as autophagy and the endo-lysosomal system. A properly functioning lysosomal system is crucial for healthy brain function, as demonstrated by rare lysosomal disorders that frequently result in fatal paediatric neurodegeneration.
While increasing the flux of material through the lysosomal system of cells in the central nervous system would clear oligomers of amyloid-β or MAPT, this only holds true while the lysosomal system is functioning well. The lysosomal system does slow down with age, but in AD it is critically damaged to the point where massive amounts of lysosomal-system cargos accumulate around amyloid plaques. This brings us to our first point, that someone who has dementia is unlikely to benefit from a therapy that enhances cargo delivery to poorly functioning lysosomes.
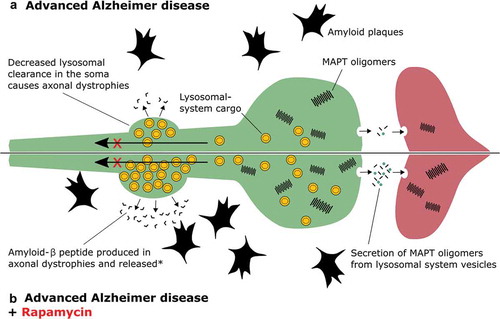
While reduced expression of lysosomal-system genes occurs naturally in the brain with aging, expression of such genes increases during AD [Citation2,Citation3]. This is presumably the brain’s response to a failing protein clearance system. This upregulation is not sufficient to rescue proteostasis in the brain, and only serves to generate more lysosomal-system cargo (such as autophagosomes), which further choke ailing lysosomes. Accumulated lysosomal-system cargo present in distended axonal dystrophies () possesses APP (amyloid beta precursor protein), as well as the proteases necessary to produce amyloid-β; in the absence of efficient lysosomal clearance, these vesicles actually produce more amyloid-β [Citation4]. Liberation of this amyloid-β can occur by cell/axonal rupture [Citation5] or by exocytosis [Citation6]. This increase in retained lysosomal-system cargo will also likely promote partial cleavage and aggregation of MAPT. These dysfunctional compartments provide a direct route of secretion to promote transcellular seeding through fusion with the plasma membrane and uptake by connected neurons [Citation7]. Treatment with rapamycin would only add fuel to this fire by increasing the rate at which lysosomal cargo is generated without fixing the underlying lysosomal dysfunction ().
Figure 1. Treatment of advanced AD with rapamycin. (A) In Alzheimer disease, lysosomal-system cargo (yellow vesicles) that should be retrogradely transported up an axon (green) to the soma, where lysosomal degradation occurs, accumulate because lysosomal function is critically damaged. (B) Treatment with rapamycin enhances the generation of lysosomal-system cargo, and during AD this will result in further accumulation of lysosomal vesicles. According to the literature this may promote propagation of MAPT aggregates to another neuron (red), and further production of amyloid plaques. *How release of amyloid-β from the dystrophy occurs is poorly understood but could happen by fusion of lysosomal-system cargo with the axon’s plasma membrane, or rupture of this membrane and release of lysosomal-system contents into the parenchyma.
This concept has been demonstrated experimentally in other model systems of neurodegenerative disease and is termed ‘autophagic stress’ – when the generation of autophagic cargo exceeds the lysosome’s capacity to clear it [Citation8]. For example, in cell models, autophagy is the mediator of neuritic pathology caused by the Parkinson disease-linked mutant of LRRK2 (leucine rich repeat kinase 2), G2019S [Citation9]. Recently, in a transgenic mouse model of amyotrophic lateral sclerosis, tissue-specific knockout of Atg7 (autophagy related 7) in motor neurons hastens disease onset. This was interpreted to mean that autophagy – which requires ATG7 – slows disease onset [Citation10]. Intriguingly, however, amyotrophic lateral sclerosis transgenic mice that cannot undergo autophagy live longer, indicating that autophagy accelerates disease at later stages [Citation10]. Genetic studies that have linked AD to sequence variation in lysosomal-system genes further support the concern that positive regulation of lysosomal-system activity late in human AD could have similar detrimental effects [Citation11].
New data from studies where amyloid plaque load was measured in people living with AD has shown that plaque deposition begins two decades before a clinical diagnosis of dementia [Citation12]. Rapamycin treatment for AD should therefore begin in a person’s 50s and would have to involve detection of early biomarkers, such as amyloid plaques with techniques such as positron-emission tomography, or promising new blood tests for amyloid-β that correlate with plaque deposition. Treatment with rapamycin at a later point would carry a higher chance of exacerbating existing lysosomal problems. The use of rapamycin early in disease development is supported by preclinical data in AD mouse models.
Preclinical studies cited by Kaeberlein and Galvan [Citation1] showed that rapamycin can reduce the accumulation of amyloid material in the brain of transgenic mice and improve cognitive function across several measures. However, except for 2 studies, the authors cite multiple investigations where rapamycin was administered to transgenic mice before the development of amyloid plaques and/or MAPT tangles. The 2 exceptions where rapamycin was administered after the beginning of amyloid plaque deposition reported a reduction in plaque burden (in mutant APPK670N,M671L,V717F- or mutant APPK670N,M671L, PSEN1S290CΔ291-319-expressing transgenic mice) [Citation13,Citation14]. Here, we point out that rapamycin treatment began in the early stage of amyloid plaque deposition, and in the absence of florid MAPT pathology. Therefore, these models resemble a person in the very early stages of Alzheimer disease.
The only study to begin rapamycin treatment both early and late in pathogenesis (in a mouse model that expressed mutant APPK670N,M671L, PSEN1M146V, and MAPTP301L) revealed that rapamycin can only prevent, but not reverse, the accumulation of amyloid plaques and MAPT tangles, as well as learning and memory deficits [Citation15]. A human who has just been diagnosed with dementia caused by AD will carry significant amyloid plaque and MAPT tangle pathology that has been developing for decades. Starting rapamycin treatment in someone with dementia will likely mimic the late intervention in Majumder et al. [Citation15], which did not work. This is consistent with our hypothesis that augmenting lysosomal-system clearance late in AD is futile, or possibly deleterious (by clogging the failing lysosomal system). This brings us to our second point that treating people with dementia is not the same as treating at-risk individuals before the onset of pathology or early in the disease process. The consensus of available preclinical data is that rapamycin could be useful to prevent the onset or early development of AD neuropathology rather than as a treatment for dementia.
Given the considerations outlined above, we propose that rapamycin should not be used in people with clinical signs of dementia. Rather, we propose that research efforts should focus on exploring prevention of amyloid plaque and MAPT tangle accumulation early in the disease process. This would involve recruiting people who are positive for amyloid plaque burden but who are still in mid-life. A trial using rapamycin in such a cohort would likely succeed in delaying signs of dementia. Trials using rapamycin in cohorts that are already affected by dementia will have a much higher chance of failure, as this drug will further damage an irreparably injured lysosomal system.
Acknowledgments
We would like to thank Prof. Christopher G Proud for reviewing this letter.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Kaeberlein M, Galvan V. Rapamycin and Alzheimer’s disease: time for a clinical trial? Sci Transl Med. 2019. DOI:10.1126/scitranslmed.aar4289
- Lipinski MM, Zheng B, Lu T, et al. Genome-wide analysis reveals mechanisms modulating autophagy in normal brain aging and in Alzheimer’s disease. Proc Natl Acad Sci USA. 2010. DOI:10.1073/pnas.1009485107
- Bordi M, Berg MJ, Mohan PS, et al. Autophagy flux in CA1 neurons of Alzheimer hippocampus: increased induction overburdens failing lysosomes to propel neuritic dystrophy. Autophagy. 2016;12:2467–2483.
- Sadleir KR, Kandalepas PC, Buggia-Prévot V, et al. Presynaptic dystrophic neurites surrounding amyloid plaques are sites of microtubule disruption, BACE1 elevation, and increased Aβ generation in Alzheimer’s disease. Acta Neuropathol. 2016. DOI:10.1007/s00401-016-1558-9
- Fiala JC. Mechanisms of amyloid plaque pathogenesis. Acta Neuropathol. 2007;114:551–571.
- Annunziata I, Patterson A, Helton D, et al. Lysosomal NEU1 deficiency affects amyloid precursor protein levels and amyloid-β secretion via deregulated lysosomal exocytosis. Nat Commun. 2013. DOI:10.1038/ncomms3734
- Polanco JC, Li C, Durisic N, et al. Exosomes taken up by neurons hijack the endosomal pathway to spread to interconnected neurons. Acta Neuropathol Commun. 2018. DOI:10.1186/s40478-018-0514-4
- Cherra SJ 3rd, Chu CT. Autophagy in neuroprotection and neurodegeneration: A question of balance. Future Neurol. 2008;3:309–323.
- Plowey ED, Cherra SJ 3rd, Liu YJ, et al. Role of autophagy in G2019S-LRRK2-associated neurite shortening in differentiated SH-SY5Y cells. J Neurochem. 2008. DOI:10.1111/j.1471-4159.2008.05217.x
- Rudnick ND, Griffey CJ, Guarnieri P, et al. Distinct roles for motor neuron autophagy early and late in the SOD1(G93A) mouse model of ALS. Proc Natl Acad Sci USA. 2017;114:E8294–E8303.
- Gao S, Casey AE, Sargeant TJ, et al. Genetic variation within endolysosomal system is associated with late-onset Alzheimer’s disease. Brain. 2018. DOI:10.1093/brain/awy197
- Roberts BR, Lind M, Wagen AZ, et al. Biochemically-defined pools of amyloid-β in sporadic Alzheimer’s disease: correlation with amyloid PET. Brain. 2017;140:1486–1498.
- Lin AL, Zheng W, Halloran JJ, et al. Chronic rapamycin restores brain vascular integrity and function through NO synthase activation and improves memory in symptomatic mice modeling Alzheimer’s disease. J Cereb Blood Flow Metab. 2013. DOI:10.1038/jcbfm.2013.82.
- Jiang T, Yu JT, Zhu XC, et al. Temsirolimus promotes autophagic clearance of amyloid-β and provides protective effects in cellular and animal models of Alzheimer’s disease. Pharmacol Res. 2014. DOI:10.1016/j.phrs.2014.02.008
- Majumder S, Richardson A, Strong R, et al. Inducing autophagy by rapamycin before, but not after, the formation of plaques and tangles ameliorates cognitive deficits. PLoS One. 2011. DOI:10.1371/journal.pone.0025416