
ABSTRACT
Steroid hormones are made from cholesterol and are essential for many developmental processes and disease conditions. The production of these hormones is nutrient dependent and tightly controlled by mechanisms that involve delivery of the precursor molecule cholesterol stored in lipid droplets (LDs). Recent studies have implicated macroautophagy/autophagy, a process regulated by nutrition, in the degradation of LDs and the mobilization of stored lipids. We recently identified an autophagy-dependent mechanism that regulates steroid production via effects on cholesterol trafficking. Through gain- and loss-of-function studies in Drosophila, we found that essential autophagy-related (Atg) genes are required in steroidogenic cells for normal steroid production. Inhibition of autophagy in these cells by knockdown of Atg genes causes strong accumulation of cholesterol in LDs and reduces steroid production, resembling effects seen in some lipid-storage disorders and steroid-dependent cancer conditions. This autophagy-dependent steroid hormone regulation (ASHR) process is regulated by the wts-yki/Warts-Yorkie tumor-suppressor pathway downstream of nutrition, coupling nutrient intake with steroid-dependent developmental growth. This mechanism potentially contributes to the development of certain cancers and lipid-storage disorders and thus may be of great therapeutic relevance.
Steroid hormones regulate a broad array of physiological processes during development and also underlie diverse disease conditions. Steroidogenesis in endocrine cells is controlled by multiple hormonal signals and regulatory mechanisms, including the controlled delivery of cholesterol, from which steroids are synthesized. These hormones are not produced and stored prior to release; rather, they are produced on demand from cholesterol stored within lipid droplets (LDs), among other structures. This observation suggests the existence of a regulatory mechanism by which LD-stored cholesterol is mobilized and trafficked for timely steroid production. However, the nature of this mechanism in endocrine steroid-producing cells, as well as its potential role in the regulation of steroidogenesis, remains poorly defined.
Because recent studies have reported the degradation of LDs by macroautophagy (hereafter autophagy), we speculated that autophagy might thus be involved in mobilizing cholesterol and that regulation of this process might be a mechanism by which cells control steroid production. It is known that autophagic activity is high in steroidogenic cells and that autophagic deficiency is associated with reduced steroid levels, but the role of autophagy in steroid hormone production remains ill-defined.
Intracellular cholesterol uptake and trafficking through the late-endosomal/lysosomal (LE/L) pathway is dependent on the NPC1 (NPC intracellular cholesterol transporter 1) protein, which mediates cholesterol transport from the LE/L compartment. NPC1 deficiency in humans causes a fatal cholesterol-storage disorder characterized by accumulation of intracellular cholesterol, and recent work has implicated autophagy in the pathogenesis of this condition. We previously found that loss of autophagy in the Drosophila steroidogenic tissue (the prothoracic gland) leads to cholesterol accumulation resembling that seen in NPC1-deficient cells. Furthermore, induction of autophagy rescues steroid-deficiency phenotypes associated with Npc1 loss in Drosophila. This suggests that autophagy may mobilize LD cholesterol for steroidogenesis and thus may be of therapeutic relevance for NPC1 disease.
To assess directly the impact of autophagy on cholesterol trafficking and steroid production, we reduced autophagy in the Drosophila prothoracic gland via RNAi against essential autophagy-related (Atg) genes (Atg1, Atg9, or Atg18). Although previous studies in other systems suggest that autophagy may be important for cholesterol uptake, we found that blocking autophagy in the prothoracic gland led to a dramatic increase in cholesterol-rich LDs, suggesting that autophagy plays a role in mobilizing cholesterol from LDs. This argues that autophagosomes traffic cholesterol, and, indeed, we found that Atg8a/LC3-positive (autophagic) vesicles sequester fluorophore-tagged cholesterol.
Because autophagy is a dynamic process in which autophagosomes undergo fusion with LE/Ls, thereby forming autolysosomes, we next investigated the nature of these cholesterol-containing autophagosomes. Through the use of a pH-sensitive fluorophore-tagged Atg8a/LC3, we found that cholesterol-containing Atg8a/LC3-positive vesicles are acidic, indicating that they are formed via autophagosome-lysosome fusion. We further found that some of these vesicles are consistently positive for late-endosomal (Rab7) and lysosomal (Lamp) markers. Interestingly, some Atg8a/LC3-positive vesicles are interconnected by tubular structures, indicating that these vesicles likely have somewhat unusual features.
To assess the potential link between autophagy and steroidogenesis, we depleted Atg expression in steroidogenic cells and found that circulating steroid titers were reduced, leading to associated developmental growth defects. Consistent with this finding, in mice, deletion of the core autophagy gene Atg5 has been linked to reduced steroid levels and steroid-deficiency phenotypes. We further found that these defects could be rescued by dietary cholesterol supplementation, indicating that cholesterol limitation underlies the steroid deficiency caused by reduced autophagy. Taken together, our findings illustrate the existence of an autophagy-mediated cholesterol-trafficking mechanism upstream of steroid synthesis (), which we have named ‘autophagy-dependent steroid hormone regulation (ASHR).’
Figure 1. A proposed model illustrating autophagy-mediated cholesterol trafficking and its role in the regulation of steroid production. In the endocrine cells of the Drosophila prothoracic gland, nutritional signaling regulates steroidogenesis by controlling autophagy-dependent mobilization of lipid droplet (LD)-stored cholesterol into the steroid-biosynthetic pathway. Atg8a/LC3-positive (autophagic) vesicles sequester and traffic the steroid hormone precursor cholesterol for delivery to the steroid biosynthetic pathway. Atg8/LC3-mediated trafficking interacts with late endosomes and lysosomes by fusion, forming amphisomes (vesicles that are positive for Atg8a/LC3 as well as Rab7, a late endosomal marker), and autolysosomes (vesicles that are positive for Atg8a/LC3 as well as a lysosomal-associated membrane protein, Lamp, a lysosomal marker). This autophagy-mediated cholesterol trafficking mechanism is controlled by nutrient-dependent (insulin signaling) activation of the wts/Warts tumor-suppressor pathway, through regulation of the microRNA ban/bantam, by its downstream effector yki/Yorkie. This process is regulated by ban through its modulatory effects on Tor and ecdysone receptor (EcR) signaling, both of which control autophagy-mediated mobilization and trafficking of LD-cholesterol.
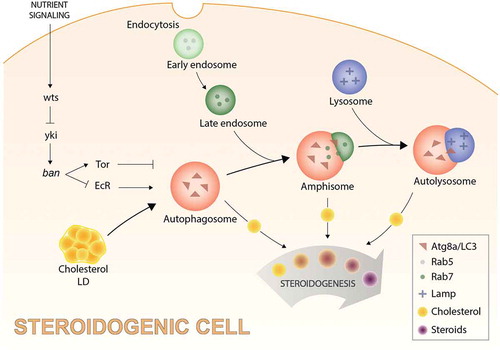
Steroid production and autophagy are nutrient-regulated processes, which argues that autophagic cholesterol trafficking should respond to extracellular levels of nutrients. We recently showed that nutrient-dependent regulation of steroidogenesis is mediated by the hpo-wts/Hippo-Warts tumor-suppressor pathway, raising the possibility that regulation of ASHR might underlie this effect. Using gain- and loss-of-function studies, we found that wts (mammalian LATS1/2) regulates autophagy through its downstream effector yki/Yorkie (mammalian YAP1), which regulates expression of the ban/bantam microRNA. Although ban has been extensively studied for its growth- and tumor-promoting effects, target genes through which this microRNA regulates growth remain elusive. Our data suggest that ban regulates growth and autophagy through effects on the Tor (Target of rapamycin) pathway, a key growth-regulatory pathway, and EcR (Ecdysone receptor), a Drosophila homolog of mammalian NR1H/liver X receptor.
NR1H is implicated in the regulation of cholesterol homeostasis, and it promotes macrophage reverse cholesterol transport, a cholesterol-efflux process that helps prevent excess cholesterol accumulation and protects against atherosclerosis. Our data suggest that EcR regulates the autophagy of LD-stored cholesterol, which may provide a new mechanism for NR1H-mediated cholesterol mobilization. Although our study shows that autophagy of cholesterol provides substrate for steroid production, it is possible that this autophagosomal mechanism for cholesterol mobilization and trafficking may also provide an alternative route for cholesterol recycling and excretion.
Macrophages that accumulate so much cholesterol that they are termed ‘foam cells’ are key factors in the formation and progression of atherosclerosis, a disease characterized by deposition of excessive cholesterol in the arterial walls. Recent work suggests that LD-stored cholesterol delivered to the lysosomal pathway via autophagy is important for reverse cholesterol transport in macrophage foam cells. We speculate that the autophagosomal cholesterol-trafficking mechanism that we have characterized in endocrine cells may be of therapeutic relevance for removing excess cholesterol via efflux or vesicle-mediated exocytosis in cholesterol-storage disorders and atherosclerosis.
It is clear that abundant intracellular cholesterol contributes to the progression of many steroid-dependent prostate and breast cancers. However, whether excess cholesterol drives carcinogenesis by facilitating de novo intratumoral steroid synthesis through increased substrate availability, or whether cholesterol itself activates growth-promoting pathways, remains unclear. Autophagy is often impaired in prostate cancers, which may, based on the role of autophagy in mobilizing LD-stored cholesterol that we have found, explain at least in part the increased LD cholesterol levels that drive cancer development. Identifying the interplay between autophagy and cholesterol metabolism in steroid-dependent cancers may provide unique opportunities for the development of new therapeutic strategies to target some of the most common and aggressive cancers.
Disclosure statement
No potential conflict of interest was reported by the authors.