
ABSTRACT
The machinery that decorates autophagic membranes with lipid-conjugated LC3/GABARAP is not yet fully understood. We recently reported the purification of the full-length ATG12–ATG5-ATG16L1 complex, and in reconstitution experiments with purified ATG7, ATG3, and LC3/GABARAP in vitro, together with rescue experiments in knockout cells, important aspects of the complete lipidation reaction were revealed. Hitherto unobserved membrane-binding regions in ATG16L1 were found, contributing to properties that explain the crucial role of this protein in membrane targeting and LC3/GABARAP lipidation in macroautophagy/autophagy and other related processes.
A key step in macroautophagy is the lipidation of LC3/GABARAP-family proteins (here collectively called ATG8), leading to the formation of ATG8-decorated phagophore double-membranes crucial for cargo recruitment and progression of the autophagic response. The lipidation cascade is dependent on the recruitment and action of several autophagy factors, where the final step is the transfer of ATG8 from ATG3 to phosphatidylethanolamine (PE) in the membrane. ATG8 becomes covalently linked to ATG3 by an activation mechanism involving the ATPase ATG7. ATG3–ATG8 then adheres to and partly inserts into the membrane through the action of an N-terminal amphipathic helix in ATG3, which orients the activated ATG8 in correct position for the transfer to the PE substrate.
The ATG12–ATG5-ATG16L1 complex is thought to mediate the lipidation reaction by recruiting ATG3–ATG8 to the membrane and to facilitate the transfer reaction to PE. How these functions are accomplished are, however, not understood. Certain proteins have been discovered that link the ATG12–ATG5-ATG16L1 complex to target membranes; the 2 best studied are the PtdIns3P-effector WIPI2 and the ULK1/2 complex subunit RB1CC1/FIP200. The role of such proteins is viewed as mediators of specificity, so that ATG8-lipidation only occurs at correct target membranes.
Due to difficulties in the isolation of a functional ATG12–ATG5-ATG16L1 complex, the investigations of this critical component have been hampered. Using a homologous expression system (HEK cells) we successfully purified the full-length hexameric complex, consisting of a dimer of ATG16L1 and conjugated ATG12–ATG5 proteins with authentic N- and C-termini (). Using this complex together with purified ATG3, ATG7, and different types of liposomes, we found a requirement for the ATG12–ATG5-ATG16L1 complex for efficient lipidation of ATG8-proteins when reconstituted in vitro. Thus, using this system, we can for the first time reconstitute the mammalian ATG8 lipidation reaction to better mimic the situation in the cell, and elucidate the detailed mechanism of the reaction.
Figure 1. Schematic depiction of ATG12–ATG5-ATG16L1 complex in solution, and bound to target membrane recruiting the ATG3–ATG8 conjugate. Regions of importance for the function of ATG16L1 are indicated. It is not known if the amphipathic α-helix (designated as ‘helix 2’) is present already in the soluble complex and interacts in the dimer (as found in crystal structures), or is formed only when the complex partially inserts into the membrane. The C terminus of ATG16L1 is thought to mediate specific membrane targeting, both through protein-protein interactions and through β-isoform-specific membrane binding.
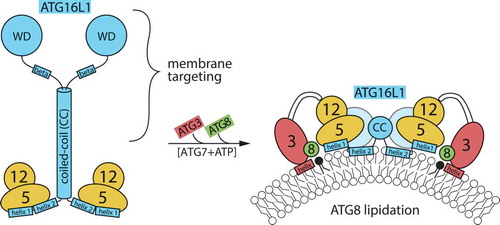
Whereas the ATG12–ATG5 conjugate alone had no tendency to adhere to membranes by itself, we found that 2 different parts in ATG16L1 have membrane-binding activity; an amphipathic helix in the N terminus and a region in the C terminus only present in the β-isoform of the protein. The N-terminal amphipathic helix (designated helix 2) is completely conserved in vertebrates and is located immediately after an ATG5-binding helix (called helix 1). We found that an intact helix 2 is required for ATG8-lipidation under all conditions tested, including canonical starvation-induced autophagy, mitophagy, LC3-associated phagocytosis (LAP), and lipidation on perturbed endosomes.
The mechanism of action of helix 2 is not yet known. We confirm other studies showing that ATG12 has a binding site for ATG3, indicating that ATG16L1 acts as a membrane-binding platform for bringing ATG3–ATG8 close to the PE substrate, via the ATG12–ATG5 conjugate. As helix 2 is situated close to the ATG5-binding helix, it can be envisioned that membrane binding by ATG16L1 brings the rest of the proteins in this machinery close enough to the membrane surface for the lipidation to occur. Here we propose 2 models for how this could be achieved: (1) The combined membrane binding affinity of the ATG3 and ATG16L1 amphipathic helices becomes sufficient for stabilization on highly curved membranes, similar to that envisioned for the leading edge of a phagophore membrane. (2) It is also possible that the assembly of the ATG12–ATG5-ATG16L1 complex into higher oligomeric structures on the surface will lead to deformations of the membrane through insertion of helix 2, so that ATG3 can insert its amphipathic helix.
Yeast and other unicellular eukaryotes express a short form of Atg16, consisting of an Atg5-binding helix, a sequence corresponding to helix 2, and a coiled-coil domain containing an interaction site for Atg21 (WIPI2 in humans), whereas metazoans have a longer form that is extended at the C terminus. It has been enigmatic why ATG16L1 in metazoans have this extension. As canonical autophagy can be fully rescued in cells by a truncated form of mammalian ATG16L1, comparable in size to yeast Atg16, additional functions of the C terminus are anticipated. In humans, 2 isoforms (designated α and β) are expressed, differing by a β-isoform-specific 19-amino acid insert. This insert contains 4 reported phosphorylation sites, highlighting it as an important regulatory region in ATG16L1. Importantly, we found that this β-insert encompasses a membrane-binding region. The β-specific membrane-binding motif is dispensable for starvation-induced autophagy, as well as mitophagy and LAP, which also proceed unaltered when the C terminus of ATG16L1 is truncated. In contrast, ATG8 lipidation on damaged endosomes is only functional when the ATG16L1 KO cells are rescued with the full-length β-isoform of ATG16L1. We also find that lipidation in vitro is enhanced by the full-length β-isoform when the reaction is performed with low-curvature membranes. Our results indicate that the membrane-binding function of the C terminus is important for ATG8 lipidation on particular membranes, and may be linked to recruitment of the lipidation machinery upon membrane events independent of the class III PtdIns3-kinase (PIK3C3) and ULK1/2 complexes. Such a function could be engaged in the defense against invading pathogens. Our finding is in line with a recent report showing that the most C-terminal part of ATG16L1, a WD-repeat β-propeller domain, is also involved in PtdIns3P-independent targeting to perturbed endosomes. In this case, however, the WD-repeat domain is suggested to facilitate targeting through a protein-protein interaction. It seems that, when needed, both protein-membrane and protein-protein interactions mediated by ATG16L1´s C-terminal part, are used to target the ATG8-lipidation machinery to cellular locations distinct from autophagic membranes in metazoans.
It is known that WIPI2 facilitates recruitment of ATG16L1 to sites of autophagosome formation. At the same time, WIPI2 is dispensable for starvation-induced autophagy. Whereas the N-terminal amphipathic helix in ATG16L1 is not important for membrane targeting in cells (which speaks more of a function in the lipidation mechanism), we found that the C terminus of ATG16L1 can compensate for the loss of WIPI2 in canonical autophagy. The result explains why knocking out WIPI2 only has a modest effect on canonical autophagy. Unexpectedly, this compensatory function is dependent on PIK3C3 activity, a finding that may indicate that the C terminus binds another, yet to-be-discovered, PtdIns3P-effector. However, our result may also be related to a recent report showing that the coiled-coil region of ATG16L1 has a binding site for PtdIns3P.
Concluding remarks: In addition to ATG16L1´s general role in the ATG8 lipidation mechanism, it facilitates recruitment of the conjugation machinery to the correct membrane site during macroautophagy and several other autophagy-related processes. Further studies are required to reveal the specificity and diversity of the protein-membrane and protein-protein interactions centered on ATG16L1 that provide target membrane specificity of the ATG8-lipidation system.
Acknowledgments
This work was partly supported by the Research Council of Norway (project number 221831) and through its Centres of Excellence funding scheme (project number 262652), as well as the Norwegian Cancer Society (project number 171318).