
ABSTRACT
The sequential action of ATG proteins guarantees the formation of the autophagosome from the steps of the induction, nucleation, elongation and sealing of the phagophore membrane. Posttranslational modifications further add to the fine-tuning regulation of this highly ordered machinery and confer, in space and time, the dynamics necessary to respond to macroautophagy/autophagy activation, and to shut it down. Recently, we reported the discovery of GAN (gigaxonin), an E3 ubiquitin ligase adaptor as a key regulator of the elongation step of phagophore formation. GAN interacts, ubiquitinates and degrades ATG16L1, which forms a complex with the ATG12–ATG5 ubiquitin-like conjugation system, and specifies the site of lipidation of LC3 by this complex onto the nascent phagophore. Accordingly, depletion of GAN in primary neurons causes the accumulation of ATG16L1 and decreases the autophagy flux by impairing the net production of autophagosomes. Considering the pivotal role of ATG16L1 in autophagy, and the reversal of the deficits upon reintroduction of GAN, one can speculate that GAN constitutes a novel molecular switch to fine tune the autophagy machinery.
Autophagy is critical for cellular and tissue homeostasis, and its dysfunction is a common determinant of numerous diseases, genetically inherited, infectious or acquired. The macroautophagy pathway (hereafter referred to as autophagy), whereby cytoplasmic components are delivered to lysosomes after engulfment of the material within the double-membrane autophagosome, is orchestrated by a complex cascade of events, implicating more than fifteen autophagy-related (ATG) proteins. ATGs act in a sequential manner to drive the induction, nucleation, elongation and sealing of the phagophore to generate the autophagosome. A higher degree of regulation of this process is achieved through post-translational modifications (PTMs), ensuring a fast and efficient response to multiple signaling sources. Thus, several hundred PTMs of ATG proteins cooperate to regulate their stability, activity, localization and interaction between each other and with numerous autophagy regulators. In this myriad of signaling events, phosphorylation represents the most extensively studied PTM and is key in autophagy induction and nucleation, but very little is known about the ubiquitin regulation of ATG proteins, and in particular the E3 ligases controlling it.
Two ubiquitin-like (UBL) conjugation systems, namely ATG12–ATG5 and LC3– phosphatidylethanolamine (a mammalian homolog of the yeast Atg8) drive the elongation step of the autophagy pathway ()). Structurally related to ubiquitin, the ATG12 and LC3–I proteins are first transferred from E1- (ATG7) to E2- (ATG10 and ATG3, respectively) like enzymes. Subsequent modifications of the first UBL system permits the conjugation of ATG12 to ATG5, whose further anchoring to ATG16L1 generates the ATG12–ATG5–ATG16L1 complex bearing the E3 ligase activity necessary to conjugate a phosphatidylethanolamine group on the LC3 protein (termed LC3-II). Targeting this E3 ligase complex to the nascent phagophore, ATG16L1 is crucial in specifying the site of lipidation. Numerous studies form yeast to mammals revealed that regardless of its alterations (overexpression or genetic ablation), ATG16L1 unbalance inevitably causes inhibition of autophagosome formation due to impaired LC3 lipidation, as a result of inefficient anchoring of ATG12–ATG5 to the phagophore. Thus, identifying modulators of ATG16L1 would represent a valuable tool to fine-tune the autophagy flux, and a potential therapeutic tool aimed at lowering autophagy activity in disease.
Figure 1. Model of action of GAN (gigaxonin) on ATG16L1 and neuronal autophagy. (a) Schematic representation of the lipidation of LC3 on membranes and action of the GAN E3 ligase on ATG16L1. (b) Current overview on neuronal autophagy (top side), illustrating the compartment-specific formation of autophagosomes (in the axonal tip and in the soma) and the impairment seen in GAN-depleted neurons (bottom side), with ATG16L1 bundling within the soma. Ub, ubiquitin; PE, phosphatidylethanolamine.
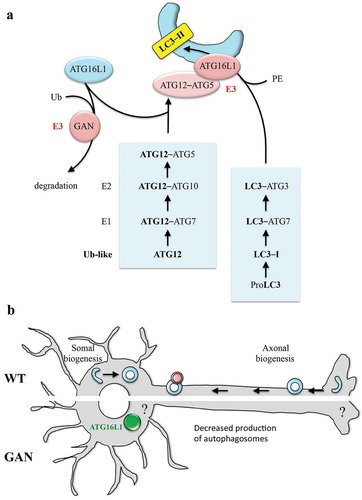
Our study [Citation1] emerged at the crossroad between the study of the signaling pathways controlled by an E3 ligase adaptor, GAN (gigaxonin), and its identification in a large study reconstructing the autophagy interaction network. The candidate approach of the possible interaction between GAN and ATG16L1 was explored to evaluate its biological relevance and its mode of regulation. In vitro experiments demonstrated that GAN interacts with ATG16L1 through its C-terminal WD40 domain, which forms a β-propeller structure prone to protein-protein interaction. Interestingly, GAN also contains a C-terminal β-propeller structure, formed of Kelch repeats. This interaction is only evidenced in the presence of a proteasome inhibitor, as the ectopic expression of GAN leads to the massive clearance of ATG16L1, overexpressed or not. While the recovery of ATG16L1 level upon inhibition of the proteasome and autophagy suggests that both degradative routes are involved in the GAN-dependent degradation, this modest effect would suggest a synergistic action, or an alternative pathway. As a result of the robust clearance of ATG16L1 by GAN, biochemical studies aimed at monitoring ATG16L1 ubiquitination are extremely challenging. Still, the indirect data of the enrichment of K48 poly-ubiquitin chains with ATG16L1 upon GAN expression is corroborated by a silencing experiment in an in vivo denaturing His-ubiquitination assay. These results permit us to speculate that GAN drives the K48 poly-ubiquitination of ATG16L1 for its degradation, but this remains to be directly demonstrated and does not exclude other (non)ubiquitin modulation of ATG16L1.
To determine the effect of the control of ATG16L1 by GAN on the autophagy flux, a novel primary neuronal model has been established, from a gan knockout mouse. Indeed, loss-of-function mutations in the GAN gene cause a severe human neurodegenerative disease named giant axonal neuropathy, which targets both the peripheral and central nervous system. Thus, in basal conditions, gan cortical neurons exhibit multiple signs of impairment of the autophagy flux, with aggregation of the autophagy receptor SQSTM1/p62 and defective autophagomose-lysosome fusion. The measure of the effectiveness of the gan neurons to respond to increased autophagy activation pinpoints a specific deficit in the net production of autophagosomes. In compliance with the action of GAN in regulating the turnover of ATG16L1, it forms ovoid bundles in the soma of gan neurons that can be resolved by ectopic expression of the human E3 ligase adaptor. Because GAN does not alter the formation of the ATG12–ATG5 elongation complex in the overexpression system, and in regards to the known role of ATG16L1 in specifying the localization of the complex to the phagophore, the speculation of an impaired anchoring of ATG12–ATG5 onto the phagophore membrane is highly plausible, but this remains to be demonstrated in a cellular system in which best resolution of individual LC3 puncta can be achieved. However, it cannot be excluded that GAN regulates ATG16L1 levels at other localizations where the recruitment of the ATG machinery is observed, such as the plasma membrane or the recycling endosomes.
For cell biology, this work places GAN (gigaxonin) as the first E3 ligase acting on the elongation step through ATG16L1. With a pivotal role in the fine-tuning of autophagosome production and other ATG16L1-dependent processes (e.g., LC3-associated phagocytosis), ATG16L1 is now manipulable by GAN, hence offering a substantial strategy for therapeutic intervention in numerous diseases with increased autophagy or autophagy-like activities. For neurobiology, the peculiar localization of ATG16L1 bundles within the soma of gan neurons opens exciting perspectives for the deciphering of the concept of autophagy compartimentalization in polarized cells ()). Indeed, in neurons, autophagosome biogenesis occurs at the distal tip of the axons, to mature and fuse with lysosomes while retrogradely transported towards the soma. Although functions of this ‘distal’ autophagy maturation have been evidenced in presynaptic and axonal homeostasis, very little is known about the ‘somal’ autophagy. Therefore, further studies on GAN will make it possible to explore this area of research on the spatial control and role of autophagy. Another interesting perspective would be to determine whether autophagy impairment, which occurs prior to degeneration in gan neurons, is a causal or contributing factor to the giant axonal neuropathy pathology. More generally, in the neurodegenerative field, GAN adds to the growing number of disease-causing proteins that regulate the autophagy pathway.
Disclosure statement
No potential conflict of interest was reported by the author.
Additional information
Funding
Reference
- Scrivo A, Codogno P, Bomont P. Gigaxonin E3 ligase governs ATG16L1 turnover to control autophagosome production. Nat Commun. 2019 Feb 15;10(1):780.