
ABSTRACT
The C-terminal domain of ATG16L1 includes 7 WD40-type repeats (WD40 domain, WDD) and is not required for canonical macroautophagy/autophagy. Instead, the WDD allows ATG16L1 to induce LC3/Atg8 lipidation in single-membrane compartments, although a detailed functional characterization of this region is still missing. In a recent report we identify the anti-inflammatory molecule TNFAIP3/A20 as a binding partner of the WDD. Such physical interaction allows mutual downregulation of the expression levels of both proteins, so that the absence of one of them causes upregulation of the other. This cross-regulation provides a molecular basis for a striking genetic interaction in mice where elimination of both molecules in the intestinal epithelium generates an aggressive inflammatory phenotype. In vitro studies reveal unexpected features of the functional interplay between ATG16L1 and TNFAIP3. ATG16L1 requires TNFAIP3 to sustain the canonical autophagic flux measured by SQSTM1/p62 degradation. The WDD mediates lysosomal degradation of TNFAIP3 promoted by ATG16L1, and also regulates the NFKB/NF-κB response. Therefore, our data reveal new roles of the WDD and TNFAIP3 in the regulation of autophagy, protein stability and inflammatory signaling. More generally, we identify the interaction between ATG16L1 and TNFAIP3 as a signaling hub that integrates different pathways with important implications for intestinal homeostasis.
Ulcerative colitis and Crohn disease are chronic inflammatory bowel diseases (IBDs) caused by the aberrant response of the host immune system to environmental factors in genetically predisposed individuals. Approximately 200 genetic variants increase the risk of developing IBD, and the proteins encoded by some of these alleles have revealed biological processes whose dysfunction may facilitate IBD. Intestinal homeostasis involves signaling pathways that prevent inflammation, autophagy and cell death, but how these signals are integrated remains unclear. In a recent publication we describe a genetic interaction between ATG16L1 and TNFAIP3/A20, a prominent anti-inflammatory regulator of NFKB activation and cell death, in IBD prevention [Citation1]. Mice harboring genetic mutations that eliminate either one of these proteins in the intestinal epithelium exhibit moderate phenotypes, but those lacking both molecules suffer a spontaneous intestinal pathology that resembles Crohn disease. Using a proteomics approach, we discovered that TNFAIP3 interacts with the WDD of ATG16L1, a binding event that involves the cooperative action of 7 WDD-binding motifs ([YFW]-X-X-[LVI]) present in the N-terminal region of TNFAIP3. Additional results suggest that the main consequence of such interaction is the mutual downregulation of both proteins, thus providing a molecular mechanism that explains their genetic epistasis. Absence of one of the partners triggers upregulation of the other, a compensatory effect that might help minimize intestinal inflammation.
But beyond the physiological implications of such crosstalk in intestinal homeostasis, our data reveal unexpected consequences of the interaction between TNFAIP3 and ATG16L1 in the regulation of autophagy. TNFAIP3-deficient mouse embryonic fibroblasts (MEFs) and intestinal organoids show increased ATG16L1, LC3-II and SQSTM1/p62 expression levels, particularly in response to TNF. Although this scenario would suggest reduced autophagic flux, treatment with bafilomycin A1 does not equalize the dissimilar LC3-II levels found in wild-type and TNFAIP3-deficient cells. This result indicates that absence of TNFAIP3 causes increased LC3-II synthesis coexisting with SQSTM1 accumulation, pointing to the intriguing idea that LC3 lipidation may be carried out ectopically, uncoupled from the canonical route that clears SQSTM1 but still resulting in the lysosomal degradation of LC3-II. The nature of such an autophagic response is unclear, but seems to have unconventional features because overexpression of the WDD represses LC3-II in a dominant-negative manner. Similar results are obtained in Tnfaip3 Atg16l1 double-deficient MEFs restored with TNFAIP3 and/or ATG16L1 constructs. Re-introduction of full-length ATG16L1 or the N-terminal domain (1-299) in this system rescues the basal LC3-II signal irrespective of whether or not TNFAIP3 is present, but SQSTM1 expression is only reduced in cells previously restored with TNFAIP3. Re-expression of ATG16L1 in TNFAIP3-deficient conditions boosts SQSTM1 levels, suggesting again that TNFAIP3 is required for proper coupling between LC3 lipidation and SQSTM1 clearance. These results raise the notion that, by interacting with the WDD, TNFAIP3 holds ATG16L1 in check to limit the activity of the LC3 lipidation complex to the canonical autophagic route that degrades SQSTM1. Because TNFAIP3 is strongly induced by TNF, and we show that TNFAIP3 binds ATG16L1 in response to TNF treatment, perhaps this mechanism operates specifically in inflammatory conditions. In the absence of TNFAIP3, unleashed ATG16L1 would induce an unconventional LC3 lipidation activity mediated by the WDD and uncoupled from SQSTM1 degradation. The nature of this response and its possible contribution to minimize intestinal inflammation in TNFAIP3-deficient conditions are questions remaining to be addressed.
We also found increased TNFAIP3 expression in Atg16l1-deficient MEFs, suggesting that ATG16L1 decreases TNFAIP3 stability. Consistently, re-introduction of full-length ATG16L1, but not the N-terminal domain, reduces the levels of TNFAIP3, indicating that ATG16L1 controls TNFAIP3 expression through the WDD. Unexpectedly, such WDD-mediated activity requires lysosomal function, because pharmacological inhibition of the lysosome tends to normalize the dissimilar TNFAIP3 levels observed in cells with different ATG16L1 status. Although a direct involvement of autophagy has not been explored, it is tempting to speculate that the WDD might target TNFAIP3 for autophagic degradation, acting in this context as an autophagy receptor.
Lastly, we found that the WDD controls the NFKB response triggered by TNF. MEFs lacking ATG16L1 respond with a higher NFKB-dependent luciferase activity to TNF treatment compared to their wild-type counterparts. Re-expression of full-length ATG16L1 represses this activity but the N-terminal domain has an incomplete effect, indicating a contribution of the WDD. How this regulation occurs is unclear, but one possibility could be that TNFAIP3 recruits ATG16L1 through the WDD to the TNF receptor signaling complex to modulate the intracellular trafficking of the signaling vesicles, thus controlling the overall signaling output.
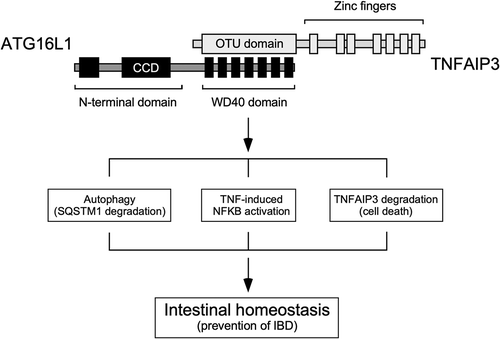
Together, our results argue that the interaction between the WDD and TNFAIP3 has a wide impact in a variety of pathways that regulate autophagy, inflammation and protein stability, and therefore it constitutes a signaling hub that integrates multiple routes to control intestinal homeostasis (). Establishing exactly how such integration takes place will require additional studies.
Figure 1. The interaction between TNFAIP3 and the WDD of ATG16L1 constitutes a signaling node that integrates pathways involved in the regulation of autophagy (SQSTM1 degradation), inflammatory signaling, protein stability and cell death, and therefore has a critical role in the maintenance of intestinal homeostasis and prevention of IBD. ATG16L1 domains: N-terminal region that suffices to sustain canonical autophagy (residues 1–299), coiled-coil domain (CCD; 78–230), WD40 repeats (WDD, 320–601). TNFAIP3 domains: ovarian tumor deubiquitinase region (OTU; 92–263), zinc fingers (381–790). The WDD and OTU domains are the minimal regions of ATG16L1 and TNFAIP3, respectively, that mediate the interaction between both proteins.
Acknowledgments
Funding for the F.X.P. lab was obtained from the Ministerio de Ciencia, Innovación y Universidades (Refs. SAF2014-53320-R and SAF2017-88390-R) of the Spanish Government, the Broad Medical Research Program and Crohn’s and Colitis Foundation (IBD-0369) and the Junta de Castilla y León local government (FIC016U14). Additional funding was received from the FEDER program of the European Union. Funding for the G.v.L. lab was obtained from the FWO, the Strategic Basic Research (SBO) programme, the Queen Elisabeth Medical Foundation, the Charcot Foundation, the Foundation against Cancer, the Cancer Research Institute Ghent (CRIG) and the “Concerted Research Actions” (GOA) of the Ghent University.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
Reference
- Slowicka K, Serramito-Gómez I, Boada-Romero E, et al. Physical and functional interaction between A20 and ATG16L1-WD40 domain in the control of intestinal homeostasis. Nat Commun. 2019;10:1834.