
ABSTRACT
We recently found that re-routing intracellular vesicle traffic by suppressing macroautophagy/autophagy or endocytosis genes drastically deregulates Drosophila intestinal stem cell (ISC) proliferation, leading to massive gut hyperplasia that has a negative impact upon lifespan. Beginning with the poorly characterized Snx (sorting nexin) genes, we surveyed a broad set of genes in the endocytosis-autophagy network and found that most of them have this effect. We then discovered that deregulated Egfr-Ras85D/Ras1-mitogen-activated protein kinase signaling is the primary trigger for ISC proliferation upon disruption of this network and determined that in the mutants, ligand-activated receptors were stabilized and recycled to the cell surface via Rab11-dependent endosomes, rather than being degraded via autophagosomes. We profiled the mutational landscape for orthologous network genes in human cancers using The Cancer Genome Atlas (TCGA), and revealed strong, novel associations with distinct genomic and epigenomic subtypes of colorectal cancer.
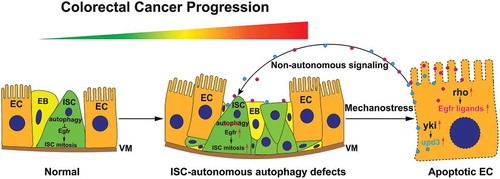
ISC-autonomous autophagy defects release stem cell proliferation
Upon gut epithelium turnover, an ISC senses different signal inputs from its surrounding niche cells, then interprets these inputs for proliferation and differentiation signaling. Aberrancies may cause colorectal cancer (CRC) or intestinal atrophy. Intracellular vesicle trafficking is known to affect signal transduction during various normal and pathophysiological processes, but how it affects the behavior of ISCs during homeostatic self-renewal of the gut is largely unexplored. While Golgi-to-ER retrograde trafficking (in particular, the COPI-Arf79F complex) was found to be required for ISC survival, the functions of intracellular vesicle trafficking in stem cells – including how autophagy affects stem cell maintenance, cancer stem cell transformation and cancer progression – remains largely unknown. Recent studies in the mouse intestine reported that autophagy genes (e.g., Atg5, Atg7, and Atg16l1) are necessary for ISC maintenance and CRC progression. However, these studies either used a pan-gut Vil1-Cre or the Paneth cell-specific Tg(Cyp1a1-cre)1Dwi/Ah-Cre, making it impossible to distinguish ISC-autonomous requirements from potential functions in other intestinal cell types such as enterocytes (ECs). Mechanistically, these papers do not pinpoint the cell types or processes underlying the described phenotypic disturbances.
Drosophila genetics uniquely provided us the ability to precisely manipulate autophagy-related genes in different cell types (e.g., ISC, enteroblast, enteroendocrine and EC cells) of the gut epithelium and investigate how autophagy regulates ISC behavior. Our studies reveal that ISC-autonomous defects in the endocytosis-autophagy network (including shi/dynamin, Rab5, Rab7, SH3PX1, Atg1, Atg5, Atg6, Atg7, Atg8a, Atg9, Atg12, Atg16 and Syx17) release ISC proliferation without regulating differentiation, potentially increasing cancer risk. We provide a detailed functional analysis of how a conserved process (autophagic flux) controls a classical growth signaling pathway (Egfr-Ras85D-mitogen-activated protein kinase), and how genes in this process (some poorly characterized, like SH3PX1) can be anti-proliferative and tumor-suppressive [Citation1].
Autophagy acts as a tumor suppressor in ISCs via Egfr degradation
Autophagy has been reported to be either tumor suppressive or oncogenic, depending upon cancer type and stage. In early carcinogenesis, autophagy can be tumor suppressive given its role in degrading oncogenic proteins and limiting genotoxic stresses that promote cancer progression. At later disease stages, autophagy can act oncogenically by providing recycled nutrients to sustain tumor metabolism. Yet, while the late stage oncogenic function of autophagy has received substantial attention in cancer studies, the early stage tumor suppressive function remains poorly explored. Whether autophagy restricts cancer stem cell transformation and cancer progression remains inconclusive. Although various studies have reported that endocytosis factors (e.g., products of dnm1, Rab5, Rab7, Rab11-OPTN/FIP2, SNX1, SNX9, and SNX16) regulate EGFR degradation, to our knowledge no findings attribute endocytosis-mediated Egfr degradation to autophagy. Importantly, no studies report effects on cell proliferation or stem cell activation, a central and functional consequence. In the context of previous literature, our discoveries that suppression of autophagy promotes Egfr stabilization, mitogen-activated protein kinase/Erk activation, and ISC proliferation, assemble a novel and crucial pathway. More recently, Kinsey et al. (2019) reported that combined inhibition of MAP2K1/MEK1-MAP2K2/MEK2 plus autophagy displays synergistic anti-proliferative effects in late stage pancreatic ductal adenocarcinoma. Based on our findings that inhibiting autophagy in early-stage tumors may trigger Egfr activation and hyperproliferation, we suggest caution in applying such strategies to early stages of carcinogenesis. Further, we also are the first to report that Rab11-dependent endosomes are required for triggering ISC division upon autophagy dysfunction. This finding suggests that genetic- or pharmacologically mediated Rab11 silencing may provide new strategies for suppressing autophagy-deficient tumors.
ISC-autonomous Egfr signaling and Ec-localized stress signaling catalyze a feed-forward loop
Upon SH3PX1-dependent autophagy loss of function, we discovered that ligand-bound Egfr will be recycled to the cell surface via Rab11-endosomes rather than degraded via autophagosomes. This hyperactivates mitogen-activated protein kinase/Erk and autonomously stimulates ISC proliferation. The excessive ISC division generates local mechano-stress and detaches surrounding ECs from the basal membrane. This triggers yki activity as well as cytokine upd3 and intramembrane protease rho (rhomboid; required for Egfr ligand cleavage and activation) production in ECs. The EC-localized stress then signals back to ISCs to promote ISC proliferation, thereby catalyzing feed-forward ISC hyperplasia (). This amplification of proliferative signaling may occur in early stage cancers.
Figure 1. The role of autophagy in regulation of intestinal stem cell proliferation and colorectal cancer progression. Blockages in the autophagy pathway stabilize Egfr, which autonomously activates ISC proliferation. Excess ISC divisions trigger general epithelial stress, EC-localized yki and rho (Egfr ligand splicer) activation, which promote the production of upd3 and the activation of Egfr ligands, respectively. This catalyzes a feedforward reaction and massive ISC hyperplasia. ISC hyperproliferation-generated local mechano-stress could potentially magnify proliferation signals. This process mimics carcinogenic processes (from early-stage cancer stem cells to cancer), and suggests a tumor suppressive role for autophagy in early stages of colorectal cancer progression. ISC, intestinal stem cell; EB, enteroblast; EC, enterocyte; VM, visceral muscle; yki, yorkie; upd3, unpaired 3; rho, rhomboid.
Clinical relevance
In cultured human cell lines, we found that repression of autophagy rapidly increases EGFR and phosphorylated MAPK/ERK levels. These data suggest that blocking autophagy can reduce the degradation of EGFR and increase downstream MAPK/ERK signaling in human cells, consistent with our findings in Drosophila ISCs. Further, using TCGA data, we demonstrate a high frequency of somatic mutations in endocytosis-autophagy network genes across multiple cancer types, suggesting functional involvement and significance. In human CRCs, we uncovered a strong association between tumors with somatic mutations in ATG and SNX genes, and microsatellite instability-high (MSI-H) genomic and CpG island methylator phenotype (CIMP) epigenomic phenotypes. We also discovered a negative association between mutations in 3 human sorting nexins (SNX9, SNX18, and SNX33, the orthologs of Drosophila SH3PX1) and activating KRAS mutations in CRC. This suggests that in humans, as in flies, deficiencies in autophagy may promote colorectal carcinogenesis by activating EGFR-MAPK/ERK signaling via a mechanism that is independent of activating mutations in EGFR, KRAS, and BRAF. Identifying how MAPK/ERK is activated in cancer stem cells that lack activating mutations in KRAS (or BRAF) will be critical toward the development of novel therapeutic strategies for CRC as well as other RAS-MAPK/ERK-dependent cancers.
Acknowledgments
This work was supported by the Huntsman Cancer Foundation and the Center for Genomic Medicine/Utah Genome Project at the University of Utah (to B.A.E. and C.M.U.) and grants from the National Institutes of Health (NIH: R01 GM124434 to B.A.E.; P30 CA042014 to B.A.E. and C.M.U.; U01 CA206110, R01 CA189184, and R01 CA211705 to C.M.U.). A.N.H. was supported by the National Human Genome Research Institute (T32 HG008962).
Disclosure statement
The authors declare no conflict of interest with this work. C.M.U. has as cancer center director oversight over research funded by several pharmaceutical companies but has not received funding directly herself.
Additional information
Funding
Reference
- Zhang P, Holowatyj AN et al. An SH3PX1-dependent endocytosis-autophagy network restrains intestinal stem cell proliferation by counteracting EGFR-ERK signaling. Dev Cell. 2019;49:574–589.e5.