
ABSTRACT
The Ser/Thr protein kinase ULK1 is an upstream macroautophagy/autophagy regulator that is rapidly activated to ensure a proper adaptive response to stress conditions. Signaling pathways modulating ULK1 activity have been extensively characterized in response to nutrient/energy shortage, which mainly act by mediating ULK1 post-translational modifications, such as phosphorylation, acetylation and ubiquitination. Less characterized is how tissue-specific stress signals are able to activate ULK1 to induce autophagy. Our recent study has uncovered the E3 ubiquitin ligase TRIM32 as a novel ULK1 activator that regulates autophagy in muscle cells upon atrophy induction. TRIM32 is conveyed to ULK1 by the autophagy cofactor AMBRA1 to stimulate its kinase activity through unanchored K63-linked polyubiquitin chains. Notably, mutations in TRIM32 responsible for limb-girdle muscular dystrophy 2H disrupt its ability to bind ULK1 and to induce autophagy in muscle cells, resulting in a dysregulated activation of the atrophic process. In conclusion, we have identified a novel molecular mechanism by which autophagy is regulated in muscles, whose alteration is associated with the development of muscular dystrophy.
Muscle atrophy is a highly regulated process aimed at reducing muscle mass during prolonged inactivity, which has to be finely tuned in order to preserve cell survival and allow tissue regrowth when activity is resumed. This process does not damage muscle cells unless abnormally induced by pathological stimuli, certain muscle gene mutations, chronic inflammation and dysmetabolic conditions. Both proteasome and autophagy machineries are activated in a complementary fashion to degrade cytoskeletal structures and organelles. The proteasome-mediated degradation requires the transcriptional activation of atrophy-specific E3 ligases, such as TRIM63/MURF1, which target sarcomeric proteins and muscle transcription factors for degradative ubiquitination. As a consequence, if the expression of TRIM63/MURF1 is inhibited, muscle atrophy is significantly reduced. In this context, autophagy is mainly involved in the degradation of organelles (e.g., sarcoplasmic reticulum and mitochondria), thus preventing harmful metabolic products, such as reactive oxygen species (ROS), to accumulate during the inactive state, which may further stimulate atrophy-specific E3 ligase expression. For this reason, when autophagy is inhibited muscle atrophy is worsened.
How atrophy stimuli are transduced to the autophagy machinery in muscle cells for a prompt activation of the process remains partly characterized. We recently found that the E3 ubiquitin ligase TRIM32 is required, both in vitro and in vivo, for the induction of autophagy in muscle cells by atrophic stimuli [Citation1].
The key role of TRIM32 in controlling muscle function initially emerged by the identification of gene mutations that are causative of limb-girdle muscular dystrophy 2H (LGMD2H). More recently, TRIM32 was shown to regulate muscle atrophy, as demonstrated in trim32 knockout mice where muscle fibers fail to regrow properly after arm immobilization.
A first hint that TRIM32 could play a role in autophagy regulation came from our observation that TRIM32 interacts with the pro-autophagic protein AMBRA1 in a proteomic screening. This interaction prompted us to analyze the role of TRIM32 in the regulation of autophagy in muscle cells. Experiments performed in myoblast cell lines showed that TRIM32 expression is not required for sustaining basal muscle autophagy, but it is essential for the induction of autophagy in response to atrophic stimuli, such as dexamethasone and nutrient starvation. Autophagy impairment was observed by measuring the degradation rate of MAP1LC3-II, SQSTM1/p62 and NBR1 by immunoblotting as well as by analyzing degradative compartments by transmission electron microscopy. We also found that autophagy inhibition in TRIM32-deficient cells results in the exacerbation of muscle damage in response to atrophic stimuli, as shown by high levels of mitochondrial ROS and increased induction of the atrophic gene TRIM63/MURF1. Importantly, we confirmed that autophagy induction is defective in muscle tissues upon dexamethasone treatment using a mouse model of Trim32 deficiency. Autophagy impairment is also observed in cells carrying LGDM2H mutants. These results were obtained using 2 independent cellular systems: a) trim32 KO C2.7 myoblasts complemented with well-characterized TRIM32 mutants (D487N and R394H), b) fibroblasts transdifferentiated to myocytes from an LGMD2H patient, carrying a complete gene deletion in one allele and a nonsense c.1837 C > T (R613X) mutation in the NHL domain of the other allele.
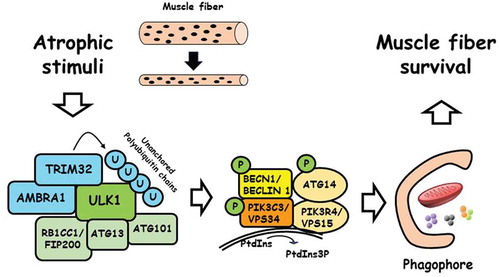
The proautophagic activity of TRIM32 relies on its ability to bind to ULK1 (). This interaction requires the presence of AMBRA1 and maps to the E3 ligase domain (RING/B-box) of TRIM32. Ectopic expression of TRIM32 promotes the activity of ULK1, as shown by monitoring BECN1/BECLIN 1, PIK3C3/VPS34 and ATG14 phosphorylation. This stimulation depends on the E3 ubiquitin ligase activity of TRIM32, which results in an apparent increase of K63-linked ubiquitination of ULK1. However, when we attempted to map the TRIM32-dependent ubiquitination sites of ULK1, we found that TRIM32 does not transfer polyubiquitin chains to ULK1 residues, but they are non-covalently bound to the carboxy-terminal domain of the protein. Interestingly, activation of protein kinases by E3 ubiquitin ligases through unchained polyubiquitin has been previously described in different signaling pathways, including the NFKB/NF-kB pathway where TRIM32 modulates in a similar manner the activity of IKBKG/NEMO.
The TRIM32-AMBRA1-ULK1 axis is relevant for the induction of autophagy in muscle cells by atrophic stimuli. In fact, we observed that, in dexamethasone-treated cells, TRIM32 is required to promote ULK1 association with K63-linked polyubiquitin chains, and increase phosphorylation of the ULK1 targets BECN1 and ATG14.
Finally, we found that pathogenic TRIM32 mutants have a strong reduction in ULK1 binding, although the interaction with AMBRA1 is not affected, and they are unable to stimulate ULK1 activity. Because ULK1 interacts with the RING/B-box domain but not with the NHL domain of TRIM32, where the pathogenic mutations are located, our results suggest that a functional NHL domain is required to make the catalytic domain of TRIM32 accessible to ULK1 upon muscle atrophy induction.
In conclusion, we have identified TRIM32 as a novel player in the regulation of ULK1 activity and we have characterized its key role in the regulation of autophagy in response to muscle atrophy. A novel aspect emerging from our study is that the ULK1 complex can be activated by unanchored polyubiquitin chains, which could represent a mechanism used by other stress stimuli, such as ubiquitinated protein aggregates and organelles, to stimulate autophagy. Moreover, these results provide novel evidence indicating that AMBRA1 acts as a determinant stress response factor to convey multiple E3 ubiquitin ligases to regulate the autophagic machinery in specific contexts, such as TRAF6 and CUL4A-CUL4B in nutrient-starvation conditions, PRKN/PARKIN and HUWE1 in response to mitochondria damage and, as described here, TRIM32 during muscle atrophy. To decipher how AMBRA1 may switch among different E3 ubiquitin ligases with little structural similarities is likely to provide relevant information on how selective autophagy is regulated.
Figure 1. The ubiquitin ligase TRIM32 regulates autophagy under atrophic conditions. In muscle fibers, TRIM32 binds AMBRA1 and ULK1 upon atrophy induction, and activates ULK1 via unanchored polyubiquitin. ULK1 triggers autophagosome formation by phosphorylating components of the BECN1/Beclin1-PIK3C3/VPS34 complex. Autophagy induction contributes to the survival of muscle fibers in atrophic conditions by ensuring mitochondria degradation and preventing ROS accumulation. P: phosphate, U: ubiquitin.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
Reference
- Di Rienzo M, Antonioli M, Fusco C, et al. Autophagy induction in atrophic muscle cells requires ULK1 activation by TRIM32 through unanchored K63-linked polyubiquitin chains. Sci Adv. 2019 May 8;5(5):eaau8857. eCollection 2019 May. PubMed PMID: 31123703; PubMed Central PMCID: PMC6527439.