
ABSTRACT
Trimethyltin chloride (TMT) is widely used as a constituent of fungicides and plastic stabilizers in the industrial and agricultural fields, and is generally acknowledged to have potent neurotoxicity, especially in the hippocampus; however, the mechanism of induction of neurotoxicity by TMT remains elusive. Herein, we exposed Neuro-2a cells to different concentrations of TMT (2, 4, and 8 μM) for 24 h. Proteomic analysis, coupled with bioinformatics analysis, revealed the important role of macroautophagy/autophagy-lysosome machinery in TMT-induced neurotoxicity. Further analysis indicated significant impairment of autophagic flux by TMT via suppressed lysosomal function, such as by inhibiting lysosomal proteolysis and changing the lysosomal pH, thereby contributing to defects in autophagic clearance and subsequently leading to nerve cell death. Mechanistically, molecular interaction networks of Ingenuity Pathway Analysis identified a downregulated molecule, KIF5A (kinesin family member 5A), as a key target in TMT-impaired autophagic flux. TMT decreased KIF5A protein expression, disrupted the interaction between KIF5A and lysosome, and impaired lysosomal axonal transport. Moreover, Kif5a overexpression restored axonal transport, increased lysosomal dysfunction, and antagonized TMT-induced neurotoxicity in vitro. Importantly, in TMT-administered mice with seizure symptoms and histomorphological injury in the hippocampus, TMT inhibited KIF5A expression in the hippocampus. Gene transfer of Kif5a enhanced autophagic clearance in the hippocampus and alleviated TMT-induced neurotoxicity in vivo. Our results are the first to demonstrate KIF5A-dependent axonal transport deficiency to cause autophagic flux impairment via disturbance of lysosomal function in TMT-induced neurotoxicity; manipulation of KIF5A may be a therapeutic approach for antagonizing TMT-induced neurotoxicity.
Abbreviations: 3-MA: 3-methyladenine; AAV: adeno-associated virus; ACTB: actin beta; AGC: automatic gain control; ATG: autophagy-related; ATP6V0D1: ATPase H+ transporting lysosomal V0 subunit D1; ATP6V1E1: ATPase H+ transporting lysosomal V1 subunit E1; CA: cornu ammonis; CQ: chloroquine; CTSB: cathepsin B; CTSD: cathepsin D; DCTN1: dynactin subunit 1; DG: dentate gyrus; DYNLL1: dynein light chain LC8-type 1; FBS: fetal bovine serum; GABARAP: GABA type A receptor-associated protein; GABARAPL1: GABA type A receptor associated protein like 1; GABARAPL2: GABA type A receptor associated protein like 2; GAPDH: glyceraldehyde-3-phosphate dehydrogenase; IPA: Ingenuity Pathway Analysis; KEGG: Kyoto Encyclopedia of Genes and Genomes; KIF5A: kinesin family member 5A; LAMP: lysosomal-associated membrane protein; MAP1LC3B/LC3B: microtubule-associated protein 1 light chain 3 beta; NBR1: NBR1 autophagy cargo receptor; OPTN: optineurin; PBS: phosphate-buffered saline; PFA: paraformaldehyde; PIK3C3/VPS34: phosphatidylinositol 3-kinase catalytic subunit type 3; PRM: parallel reaction monitoring; siRNA: small interfering RNA; SQSTM1/p62: sequestosome 1; SYP: synaptophysin; TAX1BP1: Tax1 binding protein 1; TMT: trimethyltin chloride; TUB: tubulin.
Introduction
Trimethyltin chloride (TMT) is widely used as a constituent of fungicides, plastic stabilizers in industrial and agricultural fields [Citation1], and it has been documented that TMT is detectable in foodstuffs, beverages, and household items. TMT is also generally acknowledged to have potent neurotoxic effects, with many cases of human poisoning being continuously reported over the past few decades [Citation2–5]. It selectively induces neuronal damage in both human and rodent central nervous systems (CNS), especially in the hippocampus [Citation6]. Toxicity experiments in murine have revealed TMT toxicity to depend on the dose, acute or chronic TMT administration protocols, as well as the sensitivity to epilepsy-induced brain damage [Citation7]. Dentate gyrus (DG) is susceptible to high dose exposure or in mice. However, cornu ammonis (CA) is more vulnerable to low dose exposure or in rats. Its exposure to rodents caused representative seizures and other behavioral alterations, including hyperactivity, aggression, tail mutilation, as well as impairment in learning and memory [Citation8]. Our previous research had shown SYP (synaptophysin) reduction and dopaminergic system alteration may be involved in trimethyltin-induced hippocampal damage and memory impairment in BALB/c mice [Citation9]. Despite numerous biochemical and neurochemical studies on TMT, the underlying mechanism of its neurotoxicity remains unknown.
Autophagy, a phylogenetically conserved process, is crucial for eliminating aggregated proteins and damaged organelles [Citation10]. Cytoplasmic cargo is sequestered into vesicles called autophagosomes by being engulfed by double membranes through the formation of cup-shaped structures and is subsequently delivered to acidic lysosomes for degradation [Citation11]. To maintain both cellular and metabolic homeostasis, neurons depend significantly on basal autophagy to achieve sufficient degradation of misfolded and aggregated proteins or damaged organelles [Citation12]. A defective autophagic process may be an important factor contributing to neuronal cell death in many CNS disorders, such as acute nervous system injury [Citation10], neurodegenerative diseases [Citation12], and environmental heavy metal-induced neurotoxicity [Citation13,Citation14]. Fabrizi et al. had reported TMT-impaired autophagic flux in different primary neural cells, including neurons, microglial cells, and astrocytes [Citation15–17]. Chloroquine (CQ) and 3-methyladenine (3-MA) administration worsened TMT-induced cell death while rapamycin rescued neural cells from TMT-induced toxicity [Citation15], thus suggesting autophagy may play an important role in TMT-induced neurotoxicity.
In our study, proteomics, coupled with bioinformatics analysis, revealed a critical role of the autophagy-lysosome machinery in TMT-induced neurotoxicity. MAP1LC3B/LC3B (microtubule associated protein 1 light chain 3 beta) turnover and mRFP-GFP-LC3B assay confirmed the impairment of autophagic flux by TMT, eventually contributing to Neuro-2a cell death. Notably, TMT inhibited autophagic flux by inhibiting the lysosomal proteolytic activity and altering lysosomal acidification without disrupting autophagosome formation, maturation, and autophagosome-lysosome fusion. Mechanistically, through Ingenuity Pathway Analysis (IPA), for the first time, we found KIF5A (kinesin family member 5A)-dependent axonal transport serves as a key target in the TMT-induced blockage of autophagic flux and neurotoxicity in vitro. Parallel Reaction Monitoring (PRM) further validated that overexpression of Kif5a recovered the level of autophagy-related proteins. Similarly, overexpression of Kif5a effectively alleviated TMT-induced seizures, suppressed histomorphological injury of the hippocampus, and restored autophagy flux in vivo. Our findings highlight the enhancement of KIF5A to improve autophagic flux as a promising approach against TMT-induced neurotoxicity.
Results
Autophagy plays an important role in TMT-treated Neuro-2a cells
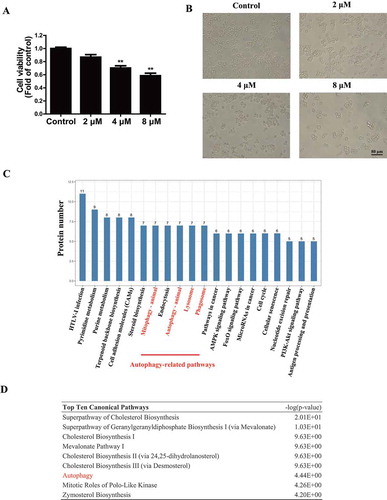
Neurotoxicity of TMT in Neuro-2a cells was determined by CCK-8 assays. Treatment of cells with 2, 4, and 8 μM TMT for 24 h caused approximately 15%, 31%, and 44% reductions in cell viability, respectively (). Moreover, after exposure to TMT, we observed a reduced number and morphological changes in Neuro-2a cells [Citation18] (). To investigate the potential molecular mechanism of TMT neurotoxicity, we determined the protein profile of the 8 μM TMT-treated group and control group using proteomic analysis. As shown in Table S1, a total of 340 proteins exhibited statistically significant changes in expression in the TMT group compared to that in the control group, with 204 proteins increased more than 1.2-fold, and 136 proteins were decreasing less than 0.8-fold. Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis investigated the pathways to determine the ones significantly affected by TMT. Interestingly, proteins in the “mitophagy,” “autophagy,” “lysosome,” and “phagosome” sets were significantly enriched in the TMT-treated group compared to that in the control group (). Moreover, autophagy was among the top 10 canonical pathways identified by IPA ( and S1). This notion reinforced the possibility that autophagy plays an important role in TMT-induced neurotoxicity.
Figure 1. Proteomics coupled with bioinformatics analysis reveals the important role of autophagy in TMT-treated Neuro-2a cells. (A) Cell viability was determined using a CCK-8 assay in Neuro-2a cells treated with TMT at different concentrations (0, 2, 4, or 8 μM) for 24 h. All experiments were repeated at least 5 times. The values are presented as means ± SEM. **P < 0.01 vs. the control group. (B) The morphology of Neuro-2a cells was observed after treatment with different concentrations (0, 2, 4, or 8 μM) of TMT for 24 h. All cells were observed with a 20 × objective. Scale bar: 80 μm. Neuro-2a cells treated with TMT (8 μM) for 24 h and control cells were harvested for proteomics, and the data were analyzed by (C) KEGG pathway enrichment analysis and (D) IPA to identify the top ten canonical pathways
TMT impairs autophagic flux in Neuro-2a cells
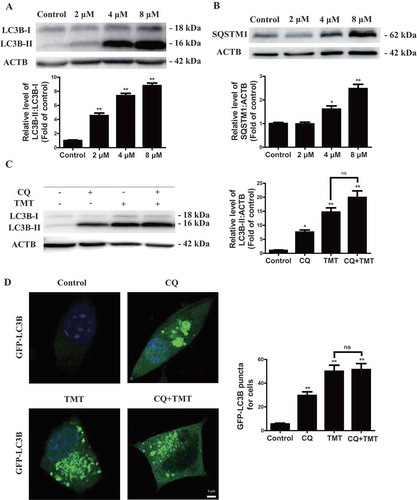
To assess the contribution of autophagy to TMT-induced neuronal cell death, we investigated the mechanism by which TMT affects autophagic flux in Neuro-2a cells. During autophagy, the cytoplasmic form LC3B-I (18 kDa) is processed and recruited to phagophores, where LC3B-II (16 kDa) is generated by lipidation at the C terminus. Thus, the amount of LC3B-II positively correlates with the number of autophagosomes. This characteristic conversion from endogenous LC3B-I to LC3B-II can be used to monitor autophagic activity [Citation19]. As shown in , TMT increased the ratio of LC3B-II:LC3B-I expression in a dose-dependent manner. Autophagy is a highly dynamic process, and autophagosome accumulation may occur due to increased autophagy induction or reduced autophagosome turnover [Citation20]. SQSTM1/p62 (sequestosome 1) is a ubiquitin-binding protein that binds LC3B-II for cargo recruitment and degradation [Citation21]. Inhibition of autophagy correlates with increased levels of SQSTM1 in mammals [Citation20]. Immunoblot analysis showed that TMT treatment increased the level of SQSTM1, which seems to confirm that TMT impairs the autophagic degradation process [Citation11] (). CQ inhibits fusion between autophagosomes and lysosomes to suppress autophagy at a late stage [Citation22]. If autophagy is induced, co-treatment with CQ will increase the LC3B-II level. On the contrary, the LC3B-II level will not be affected by the presence of CQ. TMT-induced accumulation of LC3B-II and GFP-LC3B puncta was not significantly enhanced in the presence of CQ, indicating that TMT inhibits degradation of the autophagic contents (). Together, these results provided strong evidence that TMT impaired clearance of autophagosomes, resulting in their accumulation in the cytosol. This conclusion supported TMT to be a potent autophagy inhibitor rather than an activator in Neuro-2a cells.
Figure 2. TMT inhibits autophagic flux in Neuro-2a cells. (A) and (B) Representative immunoblot and quantification analysis of LC3B and SQSTM1 in Neuro-2a cells treated with TMT at different concentrations (0, 2, 4, or 8 μM) for 24 h. ACTB was used as an internal standard for protein loading. (C) A representative immunoblot and quantification analysis of LC3B-II in Neuro-2a cells treated with TMT (8 μM) in the absence or presence of CQ (50 μM) for 24 h. ACTB was used as an internal standard for protein loading. (D) Formation of GFP-LC3B puncta in Neuro-2a cells after treatment with TMT (8 μM) in the absence or presence of CQ (50 μM) for 24 h. Scale bar: 5 μm. At least 30 cells were used to calculate the number of LC3B puncta. All experiments were repeated at least 5 times. The values are presented as means ± SEM. *P < 0.05, **P < 0.01 vs. the control group. ns means no statistical difference between the two groups
TMT does not inhibit autophagy induction or autophagosome maturation in Neuro-2a cells
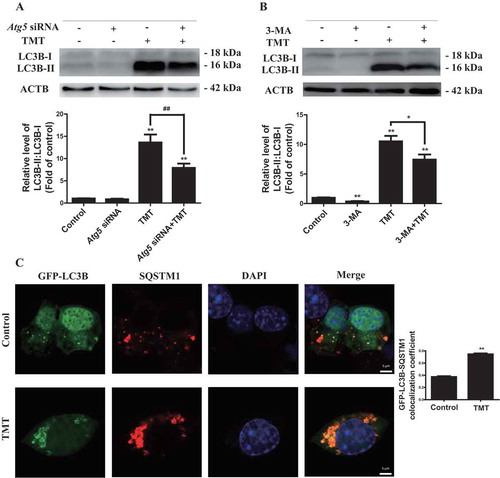
Since autophagy is a multistep process, we investigated whether TMT affects it at any stage prior to its induction. The phagophores formed in cells are dynamic and dependent on ATG5 (autophagy related 5) conjugation to ATG12 (autophagy related 12) and PIK3C3/VPS34 (phosphatidylinositol 3-kinase catalytic subunit type 3) kinase activity [Citation23]. We transfected Neuro-2a cells with Atg5 small interfering RNA (siRNA) in the presence or absence of TMT. Atg5 siRNA effectively decreased the level of ATG5 protein, as shown in Figure S2. Here, Atg5 knockdown or autophagy inhibitor 3-MA treatment did reduce the accumulation of LC3B-II under TMT treatment, indicating that TMT has no obvious effect on phagophore formation ().
Concurrent with autophagosome maturation, cargo incorporation occurs. Incorporation of one specific cargo, polyubiquitinated protein aggregates, is mediated by the receptor protein SQSTM1 [Citation24]. Cargo incorporation, which we evaluated by the colocalization of GFP-LC3B and SQSTM1, occurs during autophagosome maturation [Citation20]. Unlike the control treatment, TMT also led to SQSTM1 and GFP-LC3B puncta accumulation with more extensive colocalization ( and S3A). These data indicated that TMT does not block the cargo incorporation process associated with autophagosome maturation.
Figure 3. TMT does not affect the induction stage of autophagy or autophagosomal maturation in Neuro-2a cells. (A) A representative immunoblot and quantification analysis of LC3B in Neuro-2a cells treated with TMT following transfection with 100 nM of Atg5 siRNA or a control siRNA. (B) A representative immunoblot and quantification analysis of LC3B in Neuro-2a cells treated with TMT in the absence or presence of 3-MA (2 mM) for 24 h. (C) Immunofluorescence analysis of Neuro-2a cells expressing GFP-LC3B with an anti-SQSTM1 antibody after TMT (8 μM) treatment for 24 h. Scale bar: 2 μm. Colocalization coefficient of GFP-LC3B puncta and SQSTM1 puncta were calculated using 30 cells. All experiments were repeated at least 3 times. The values are presented as means ± SEM. **P < 0.01 vs. the control group. ##P < 0.01 vs. TMT group
TMT does not affect autophagosome-lysosome fusion in Neuro-2a cells
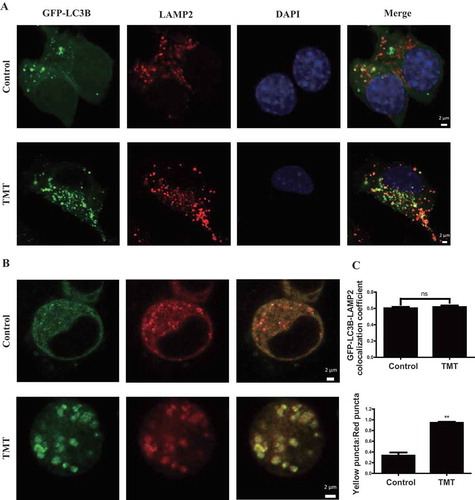
Fusion of autophagosomes and lysosomes is essential for autophagic degradation. Decreased colocalization of the autophagosome marker GFP-LC3B with the lysosomal marker LAMP2 (lysosomal associated membrane protein 2) would suggest reduced fusion; however, this was not observed in TMT-treated cells ( and S4B). RFP-GFP-LC3B is another tool used to detect fusion; GFP fluorescence is quenched when an acidic environment is encountered. Typically, yellow puncta represent pre-fusion autophagosomes, and red puncta represent post-fusion autophagosomes [Citation14,Citation20]. Since TMT significantly increased the ratio of yellow to red puncta ( and S3B), if fusion indeed occurred, as suggested by , then lysosomes are apparently less acidic.
Figure 4. TMT does not affect autophagosome-lysosome fusion in Neuro-2a cells. (A) Immunofluorescence analysis of Neuro-2a cells expressing GFP-LC3B with an anti-LAMP2 antibody after TMT (8 μM) treatment for 24 h. Scale bar: 2 μm. (B) Immunofluorescence analysis of Neuro-2a cells after treatment with TMT (8 μM) for 24 h following transfection with RFP-GFP-LC3B. Scale bar: 2 μm. (C) Colocalization coefficient of GFP-LC3B puncta and LAMP2 puncta, and the ratio of yellow puncta and red puncta following the transfection of Neuro-2a cells with RFP-GFP-LC3B after treatment with TMT (8 μM) for 24 h. At least 30 cells were used to calculate the results. The values are presented as means ± SEM. *P < 0.05, **P < 0.01 vs. the control group. ns means no statistical difference between the 2 groups
TMT inhibits lysosomal functions in Neuro-2a cells
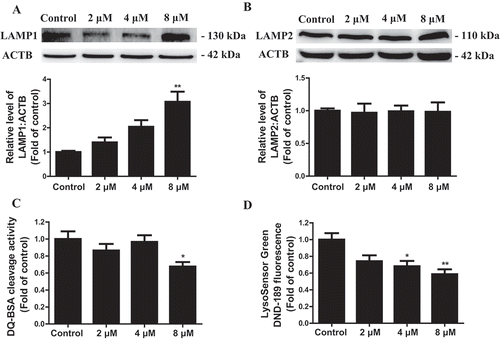
To determine whether TMT suppresses lysosomal functions, we detected the levels of LAMP1 (lysosomal associated membrane protein 1) and LAMP2 marker proteins distributed on the surface of the lysosomal membrane. While TMT increased the level of LAMP1, it had no effect on LAMP2 (), thus suggesting accumulated lysosomes to be observed in the TMT-treated Neuro-2a cells. Next, we assessed intracellular proteolytic activity. DQ™ Red BSA is heavily labeled with a strong self-quenching fluorescent BODIPY dye; proteolysis of the BSA conjugates results in the dequenching of the released protein fragments, and hence, fluorescence intensity is positively correlated with proteolytic ability [Citation25]. Intracellular proteolytic activity was obviously inhibited in Neuro-2a cells by different concentrations of TMT treatment over 24 h (). LysoSensor Green DND-189 dye is an acidotropic probe that appears to accumulate in acidic organelles, such as lysosomes, and exhibits a pH-dependent increase in fluorescence intensity upon acidification [Citation26]. TMT reduced the LysoSensor Green DND-189 fluorescence in a dose-dependent manner (), thereby demonstrating that it altered lysosomal pH in Neuro-2a cells. Together, the above results indicated impairment of autophagic flux by TMT by inhibiting lysosomal functions in Neuro-2a cells.
Figure 5. TMT impairs lysosomal function in Neuro-2a cells. (A) and (B) Representative immunoblots and quantification analysis of LAMP1 and LAMP2 in Neuro-2a cells treated with TMT at different concentrations (0, 2, 4, or 8 μM) for 24 h. ACTB was used as the internal standard for protein loading. (C) DQ-BSA staining fluorescence intensity was used to analyze lysosomal protease activity in Neuro-2a cells treated with TMT at different concentrations (0, 2, 4, or 8 μM) for 24 h. (D) LysoSensor DND-189 fluorescence intensity in Neuro-2a cells treated with TMT at different concentrations (0, 2, 4, or 8 μM) for 24 h. All experiments were repeated at least 5 times. The values are presented as means ± SEM. *P < 0.05, **P < 0.01 vs. the control group
KIF5A may be a key protein involved in regulating TMT-impaired autophagic flux in Neuro-2a cells
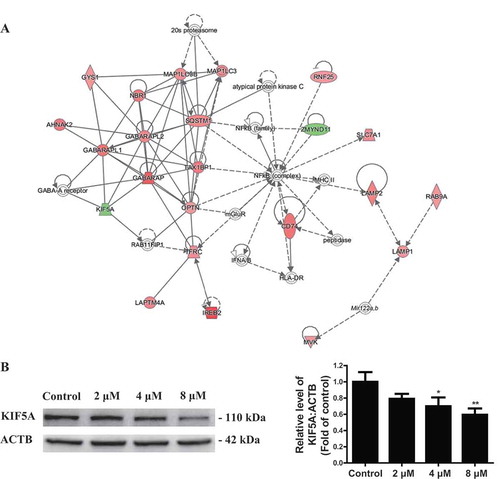
To investigate the potential molecular mechanism of TMT-impaired autophagic flux, we analyzed the molecular interaction network by IPA. An important protein integrated network was “Cell Morphology, Cellular Function and Maintenance, Cellular Compromise,” with a Z score of 34. A total of 22 proteins, including LC3/Atg8 family members such as MAP1LC3B, GABARAP (GABA type A receptor-associated protein), GABARAPL1 (GABA type A receptor associated protein like 1) and GABARAPL2 (GABA type A receptor associated protein like 2) and autophagy receptor proteins such as SQSTM1, TAX1BP1 (Tax1 binding protein 1), OPTN (optineurin), and NBR1 (NBR1 autophagy cargo receptor), were involved in this autophagy-related network (, Table S2). Notably, IPA showed the inhibition of KIF5A signaling pathway to significantly correlate with autophagy-related proteins, and KIF5A to be an important regulator of autophagic flux by interaction with GABARAP, GABARAPL1, and GABARAPL2 (). Moreover, western blotting analysis revealed that KIF5A expression was reduced in a dose-dependent manner in the TMT-treated Neuro-2a cells (). Hence, we hypothesized that KIF5A might act as a critical factor in TMT-impaired autophagic flux.
Figure 6. IPA identifies a key protein, KIF5A, in TMT-treated Neuro-2a cells. (A) The molecular interaction network “Cell morphology, cellular function and maintenance, cellular compromise” identified by IPA. (B) A representative immunoblot and quantification analysis of KIF5A in Neuro-2a cells treated with TMT at different concentrations (0, 2, 4, or 8 μM) for 24 h. ACTB was used as an internal standard for protein loading. The experiments were repeated at least 3 times. The values are presented as means ± SEM. *P < 0.05, **P < 0.01 vs. the control group
Kif5a overexpression prevents TMT-induced neurotoxicity and autophagic flux impairment in Neuro-2a cells
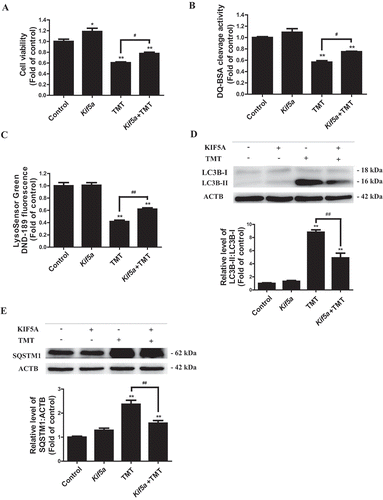
To further confirm the potential role of KIF5A in TMT-impaired autophagic flux, we overexpressed Kif5a in Neuro-2a cells (Figure S5A). As expected, Kif5a overexpression obviously restored cell viability in 8 μM TMT-treated Neuro-2a cells ( and Table S6). More importantly, we detected an improvement in proteolytic ability and restoration of an acidic environment ( Table S7, and S8), suggesting the overexpression of Kif5a to rescue lysosomal function in Neuro-2a cells. Subsequently, the levels of LC3B-II and SQSTM1 were also remarkably reduced by Kif5a overexpression (). CQ treatment could successfully block the effect of Kif5a overexpression on LC3B-II (Figure S6).
Figure 7. Kif5a overexpression prevents TMT-induced neurotoxicity and autophagic flux impairment in Neuro-2a cells. (A) Cell viability, (B) DQ-BSA staining fluorescence intensity, and (C) LysoSensor DND-189 fluorescence intensity were determined in Neuro-2a cells treated with TMT (8 μM) following transfection with 0.8 μg/mL Kif5a plasmid or a control plasmid for 24 h. Representative immunoblots and quantification analysis of (D) LC3B and (E) SQSTM1 in Neuro-2a cells treated with TMT (8 μM) following transfection with a Kif5a plasmid or a control plasmid for 24 h. ACTB was used as an internal standard for protein loading. All experiments were repeated at least 5 times. The values are presented as means ± SEM. *P < 0.05, **P < 0.01 vs. the control group. #P < 0.05, ##P < 0.01 vs TMT group
PRM is usually applied for proteomic data verification; thus, we used PRM-based targeted proteomics to confirm whether Kif5a overexpression produces any reversal of autophagy-related proteomics. Although PRM could not steadily recognize MAP1LC3B and NBR1 due to a lack of unique peptides, overexpression of Kif5a led to a significant decrease in the level of SQSTM1 (Figure S7A, Table S3). Consistently, the degradation of TAX1BP1, GABARAPL2, and OPTN was also enhanced by Kif5a overexpression (Figure S7b–D, Table S3). Interestingly, overexpression of Kif5a had no significant effect on LAMP1 expression (Figure S7E, Table S3). Together, these results demonstrated the protection of Neuro-2a cells from TMT-induced neurotoxicity due to Kif5a overexpression via the maintenance of autophagic flux.
Kif5a overexpression restores axonal transport in primary hippocampal neurons
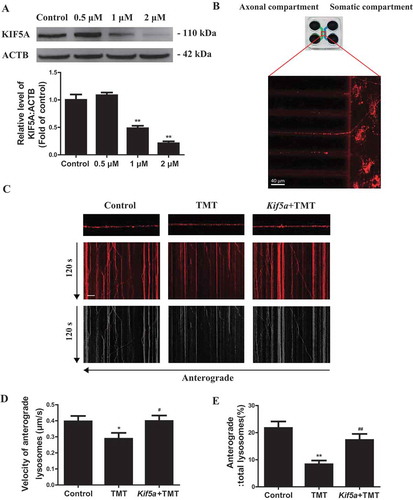
To further investigate the mechanism by which KIF5A reduction could induce lysosomal dysfunction, we first detected the interaction between KIF5A and LAMP2. TMT significantly disrupted the interaction between KIF5A and LAMP2 in Neuro-2a cells (Figure S8). KIF5A is microtubule-dependent molecular motors for intracellular transport and had been reported to transport various types of cargo, including lysosome, in the neurons of mammalian nervous systems [Citation27]. Lysosome moves bi-directionally along microtubule tracks between the center and periphery of neurons, and its positioning and motility influence multiple functions [Citation28]. Hence, we detected the axonal transport of lysosomes in primary hippocampal neurons. TMT significantly reduced cell viability of primary hippocampal neurons (Figure S9) and inhibited KIF5A expression (). Next, we observed axonal transport via live imaging of isolated hippocampal neurons grown on microfluidic chambers (), which separated axons from dendrites [Citation29]. TMT decreased the average anterograde transport velocities and anterograde transport percent of lysosomes. Importantly, Ad/Kif5a effectively rescued the TMT-induced axonal transport defects (). Together, KIF5A-dependent axonal transport defects may be involved in TMT-induced lysosomal dysfunction and autophagic flux impairment.
Figure 8. Kif5a overexpression restores axonal transport in primary hippocampal neurons. (A) A representative immunoblot and quantification analysis of KIF5A in primary hippocampal neurons treated with TMT at different concentrations (0, 0.5, 1, or 2 μM) for 24 h. ACTB was used as an internal standard for protein loading. All experiments were repeated at least 5 times. (B) Schematic of the microfluidic chamber where primary hippocampal neurons were plated on the somatic compartment and extend their axons into the axonal compartment. Neurons were labeled with LysoTracker™ Red DND-99. Scar bar: 40 μm. (C) Representative kymographs for movement of LysoTracker™ Red DND-99 puncta along axons in primary hippocampal neurons treated with TMT (2 μM) for 24 h following transfection with an Ad/Kif5a or an Ad/control. Scale bars: 10 μm and 120 s. (D) Average anterograde transport velocity of movable lysosomes (μm/s) and (E) percentages of anterograde-transported lysosomes were calculated in primary hippocampal neurons treated with TMT (2 μM) for 24 h following transfection with an Ad/Kif5a or an Ad/control. At least 8 neurons were used in each group in this experiment. The values are presented as means ± SEM. *P< 0.01 vs. the control group. #P< 0.05, ##P< 0.01 vs TMT group
Kif5a deficiency causes TMT-impaired autophagic flux in vivo
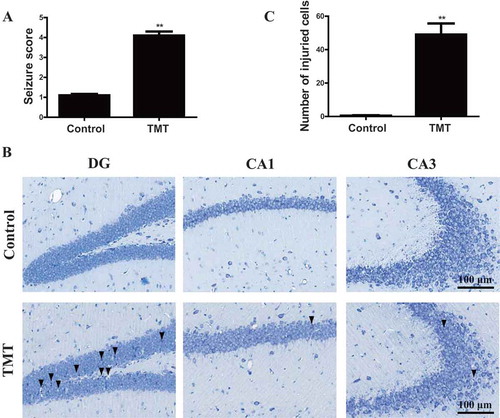
We next determined whether KIF5A deficiency causes TMT-impaired autophagic flux in vivo. C57BL/6 J mice received a single intraperitoneal injection of 2.8 mg/kg TMT for 24 h. Seizure is a representative symptom of the acute phase of TMT exposure in mice [Citation8]. In the TMT-administered mice, seizure score was significantly elevated than the controls (). The hippocampus is recognized as the main target of TMT neurotoxicity. Nissl staining of the hippocampus was performed for histological observation. As shown in , neurons in the control group were regular and intact, with abundant cytoplasm and Nissl bodies. In the TMT group, greatest changes were observed in the DG region, including neuronal cell loss, nuclear shrinkage, and dark staining of neurons [Citation30]. In addition, the neurons in the CA1 and CA3 regions exhibited loss of Nissl staining compared to that in the control group. Some neurons had blurred cell outlines, atrophic cell bodies, and vacuolization, thus revealing TMT-induced neuronal damage in the hippocampus ().
Figure 9. TMT induces neurotoxicity in C57BL/6 J mice. (A) Seizure scores were determined after the mice were administered (i.p.) 2.8 mg/kg TMT for 24 h. We observed 20 mice in each group. The values are presented as means ± SEM. **P <.01 vs. the control group. (B) Nissl staining was performed on mouse brains treated with 2.8 mg/kg TMT for 24 h. The DG, CA1, and CA3 regions of the hippocampus were observed to evaluate TMT neurotoxicity. (▼) labeled in DG exhibited neuronal cell loss, nuclear shrinkage, and many dark staining of neurons. (▼) labeled in CA1 exhibited neuronal cell loss. (▼) labeled in CA3 exhibited loss of Nissl body. We observed 4 mice in each group. All the sections were observed with a 20 × objective. Scale bar: 100 μm. (C) The number of injured cells in the DG, CA1, and CA3 regions of the hippocampus. We used 4 mice to count in each group. The values are presented as means ± SEM. **P< 0.01 vs. the control group
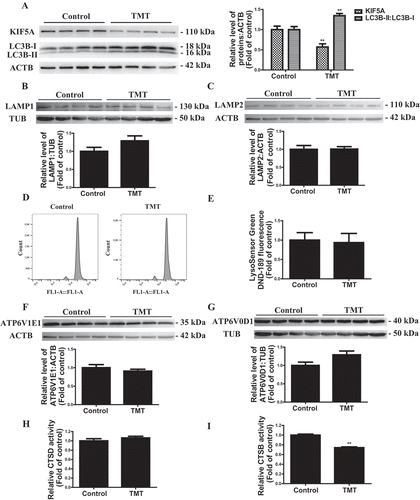
Notably, immunofluorescence and western blotting analysis of the TMT-treated hippocampus showed a block of autophagy, evidenced by the remarkable accumulation of LC3B-II and downregulation of KIF5A ( and S10). Moreover, the levels of LAMP1 and LAMP2 were not significantly different between the control group and the TMT group (). In addition, we screened the mRNA levels of the vacuolar H+-ATPase family, which is important for maintaining an acidic environment of the lysosome [Citation31]. Of all the vacuolar H+-ATPases expressed in the hippocampus, Atp6v1e1 (ATPase H+ transporting lysosomal V1 subunit E1) and Atp6v0d1 (ATPase H+ transporting lysosomal V0 subunit D1) exhibited significantly changed mRNA levels after TMT administration (Figure S11). However, there was no obvious change at the protein level (), as well as intralysosomal pH in the hippocampus (), and these results demonstrated that vacuolar H+-ATPases are not involved in TMT-associated lysosomal dysfunction in vivo. CTSB (cathepsin B) activity was dramatically decreased in the TMT-treated mice, although there was no obvious change in the activity of CTSD (cathepsin D) (), hence confirming a dramatic impairment of lysosomal protease activity.
Figure 10. TMT induces a reduction in KIF5A, and the accumulation of LC3B and impaired lysosomal function in the hippocampus of C57BL/6 J mice. (A) Representative immunoblots and quantification analysis of hippocampal KIF5A and LC3B in mice administered (i.p.) 2.8 mg/kg TMT for 24 h. ACTB was used as an internal standard for protein loading. (B) and (C) Representative immunoblots and quantification analysis of hippocampal LAMP1 and LAMP2 in mice administered (i.p.) 2.8 mg/kg TMT for 24 h. TUB and ACTB were used as internal standards for protein loading. (D) and (E) Flow cytometry analysis the LysoSensor DND-189 fluorescence intensity in the hippocampal neurons of the mice administered (i.p.) 2.8 mg/kg TMT for 24 h. (F) and (G) Representative immunoblots and quantification analysis of ATP6V1E1 and ATP6V0D1 in mice administered (i.p.) 2.8 mg/kg TMT for 24 h. TUB (from the same membrane as LAMP1 above) and ACTB (from the same membrane as LAMP2 above) were used as internal standards for protein loading. Eight mice were used in each group in the above experiments. Hippocampal (H) CTSD activity and (I) CTSB activity was detected in mice administered (i.p.) 2.8 mg/kg TMT for 24 h. The activity was normalized to the protein concentration of the samples. We used 10 mice in each group in this experiment. The values are presented as means ± SEM. **P < 0.01 vs. the control group
Kif5a overexpression antagonizes TMT-induced neurotoxicity and autophagic flux impairment in vivo
To further investigate whether KIF5A protects against TMT-induced neurotoxicity in vivo, adeno-associated virus (AAV)-Kif5a was constructed and delivered to mice by tail vein injection. As expected, Kif5a overexpression effectively restored the level of KIF5A in the TMT group (, S5, and S12). Kif5a-overexpressing mice obviously alleviated seizure symptoms ( and Movie S1-3) and hippocampal injury () compared to TMT group mice. Meanwhile, Kif5a overexpression also reduced the accumulation of LC3B-II ( and S12) and increased CTSB activity in the hippocampus (). Together, the overexpression of KIF5A alleviated TMT-induced neurotoxicity by restoring autophagic flux in vivo.
Figure 11. Overexpression of KIF5A protects against TMT neurotoxicity in vivo. (A) Representative immunoblots and quantification analysis of hippocampal KIF5A in Kif5a-overexpressing mice administered (i.p.) 2.8 mg/kg TMT for 24 h. ACTB was used as an internal standard for protein loading. We used 9 mice in each group in this experiment. (B) Seizure scores were determined in Kif5a-overexpressing mice administered (i.p.) 2.8 mg/kg TMT for 24 h. We observed 20 mice in each group. (C) Nissl staining in the DG, CA1, and CA3 regions of the hippocampus were performed in the brains of Kif5a-overexpressing mice administered 2.8 mg/kg TMT for 24 h. We observed 4 mice in each group. In the AAV-null + TMT group, (▼) labeled in DG exhibited neuronal cell loss, nuclear shrinkage, and dark staining of neurons. (▼) labeled in CA1 exhibited nuclear shrinkage. (▼) labeled in CA3 exhibited nuclear shrinkage and vacuolization. In AAV-Kif5a+TMT, (▼) labeled in DG exhibited neuronal cell loss and nuclear shrinkage dark staining of neurons. All sections were observed with a 20 × objective. Scale bar: 100 μm. (D) The number of injured cells in the DG, CA1, and CA3 regions of the hippocampus. We used 4 mice to count in each group. The values are presented as means ± SEM. *P< 0.05, **P< 0.01 vs. the control group. ##P< 0.01 vs. the TMT group (E) A representative immunoblot and quantification analysis of hippocampal LC3B in mice administered (i.p.) 2.8 mg/kg TMT for 24 h. ACTB was used as an internal standard for protein loading. We used 9 mice in each group in this experiment. (F) Hippocampal CTSB activity was detected in Kif5a-overexpressing mice administered (i.p.) 2.8 mg/kg TMT for 24 h. The activity was normalized to the protein concentration of the samples. We used 9 mice in each group in all experiments. The values are presented as means ± SEM. *P < 0.05, **P < 0.01 vs. the control group. #P < 0.05, ##P < 0.01 vs TMT group
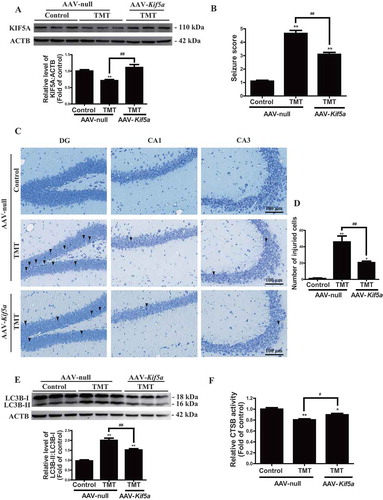
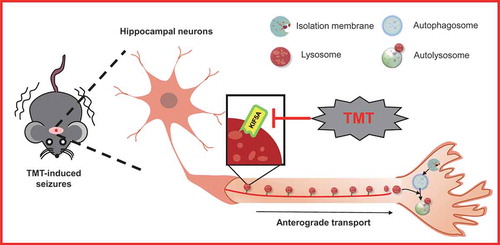
Figure 12. Schematic model of autophagic flux impairment caused by KIF5A-dependent axonal transport deficiency in TMT-induced neurotoxicity
Discussion
TMT is a byproduct of organotin compounds, which are dominant polyvinyl chloride stabilizers. These stabilizers are utilized in the United States, and >15,000 tons are used annually in Europe [Citation32]. In China, due to the wide industrial applications of organotin compounds, new patients with TMT poisoning are continuously reported [Citation33]. Workers exposed to TMT exhibit urine levels of TMT up to 3.75–13.31 μM [Citation34]. Humans occupationally exposed to TMT develop a syndrome characterized by disorientation, amnesia, hearing loss, aggressive behavior, complex partial and tonic-clonic seizures, nystagmus, ataxia, and mild sensory neuropathy [Citation2,Citation4,Citation35]. The limited reports limbic system damage in humans appears to overlap quite well with those in rodents [Citation36]. Several studies have indicated that TMT-induced neuronal injury is mainly due to mitochondrial dysfunction, Ca2+ overload, or neuroinflammation [Citation6]. However, based on proteomics coupled with bioinformatics analysis, our results are the first to demonstrate that: (i) autophagy plays an important role in TMT-induced neurotoxicity; (ii) TMT impairs autophagic flux via inhibiting lysosomal function, specifically suppressing proteolytic activity and changing the lysosomal pH; and (iii) KIF5A-dependent axonal transport is involved in TMT-induced neurotoxicity and lysosomal dysfunction both in vitro and in vivo. Our results provide new insight into TMT-induced neurotoxicity.
Previous studies had indicated, based on immunoblotting analysis and biochemical tests, the expression of several important proteins and activities of some enzymes are significantly altered in response to TMT treatment. Mass spectrometry-based proteomics is a broadly effective method for the identification, characterization, and quantification of proteins that directly perform biological functions in cells [Citation37]. A total of 340 proteins exhibited a ≥ 1.2-fold change at the protein level between the control group and the TMT group. Multidimensional analysis of the proteome sometimes allows us to deeply understand various cellular and physiological processes [Citation38]. In our study, analysis with KEGG, which is one of the databases commonly used in pathway research, demonstrated that a large number of proteins are involved in autophagy after TMT exposure. Moreover, IPA, which is a tool that uses the most comprehensive and well-documented biochemical analysis database, also confirmed autophagy-lysosome-associated pathways to be significantly affected in TMT-treated Neuro-2a cells. Interestingly, microarray-based, genome-wide expression analysis was used earlier to investigate the molecular changes, including neuroinflammation, intracellular Ca2+ overload, and oxidative stress, occurring in the TMT-injured hippocampus [Citation39]. Our study, based on proteomics coupled with bioinformatics analysis, provided new evidence for the important role of autophagy in TMT-induced neurotoxicity.
Neuro-2a cell is a widely used nervous cell line that explores the mechanism underlying neurological diseases, such as heavy metal-induced neurotoxicity [Citation14], neurodegenerative diseases [Citation40], biotoxin screening [Citation41], and neurite extension [Citation42]. Moreover, Neuro-2a was also used to investigate the autophagy-associated mechanisms in CNS, including neurodegenerative diseases [Citation43], melatonin pharmacology [Citation14,Citation44], and chronic cerebral hypoperfusion [Citation45]. Hence, Neuro-2a was applied in our study to test whether autophagy is involved in TMT-induced neurotoxicity. Autophagy is a highly dynamic process in which cytoplasmic cargo targeted for degradation is enclosed in autophagosomes [Citation46]. In the CNS, neurons require high levels of energy to perform their functions, and transport proteins, organelles, and autophagosomes for intercellular communication with specialized structures (axons, dendrites, and synapses). Defects in autophagic pathways may affect energy metabolism and intercellular communication, hence contributing to neurotoxicity [Citation13]. Recently, TMT has been described to induce the accumulation of autophagic vacuoles in the rat brain; similar vesicles have also been found in exposed (poisoned) human brains [Citation16]. Consistent with these previous studies, our results also showed blockage of autophagic flux by TMT in Neuro-2a cells. Autophagic flux is a multistage process that includes the induction stage, in which the phagophore forms, the maturation stage, in which the phagophore expands into an autophagosome, the fusion stage, in which the autophagosome fuses with the lysosome, and a final degradation stage in the lysosome [Citation11]. In our study, we found TMT did not affect autophagy induction, autophagosome maturation, or autophagosome fusion with lysosomes but significantly impaired lysosomal function. Hence, lysosomal dysfunction is the key reason for TMT-induced autophagy impairment.
Lysosomes are known as terminal degradation stations and respond dynamically to many cellular processes, especially in autophagy [Citation47]. Lysosomal dysfunction due to abnormal soluble lysosomal hydrolases, membrane proteins, or lysosomal accessory proteins has been associated with neuropathology, especially in lysosomal storage disorders and many other CNS diseases [Citation48]. Accumulation of lysosome-like structures or acidic vacuoles, as well as the increased expression of LAMP1, have been reported in TMT neurotoxicity [Citation15,Citation49,Citation50]. We also observed an increase in the level of LAMP1, further confirming injured nerve cells to contain accumulations of lysosomes and lysosome-like structures. Moreover, lysosomal function largely depends on lysosomal hydrolase activity and the acidic environment of the lysosomal lumen [Citation51]. The most important biochemical feature of lysosomes is their acidic lumen (pH 4.5 ~ 5.0), which contains >50 acidic hydrolases and lipases designated for all types of macromolecules [Citation52]. Results of the present study show impairment of lysosomal function by TMT via inhibition of lysosomal proteolysis and alteration of lysosomal pH. Consequently, the lysosome is a critical target of TMT insult.
The IPA-identified autophagy-related network, affected most significantly by TMT, showed decreased input through KIF5A, a microtubule-dependent molecular motor that belongs to the kinesin superfamily of proteins and has been reported to transport various types of cargo in the neurons of the mammalian nervous system [Citation27,Citation53]. KIF5A is established to be involved in the transport of lysosomes, synaptic vesicle precursors, synaptic membrane precursors, other diverse vesicles, and mitochondria [Citation27]. Recently, Blakeslee WW group reported KIF5A to regulate the intracellular transport of cargo, such as autophagosomes, in cardiomyocytes [Citation54]. Our results demonstrated an obvious reduction of KIF5A expression TMT, and obvious reduction of TMT-induced lysosomal dysfunction and autophagic flux impairment by overexpression of KIF5A. However, the mechanism by which KIF5A reduction could promote lysosomal dysfunction has not yet been clearly elucidated.
Neurons are extremely polarized cells consisting of a small cell body, branched dendrites, and an extended long axon. Axonal transport is fundamental for neuronal development and maintenance of effective neuronal function in mature cells, owing to the extreme polarity and size of these cells [Citation55]. The axon depends on the biosynthetic and degradative activities of the soma and participates in intracellular neural transmission for responding effectively to trophic signals or stress insults [Citation56]. The degradative housekeeping functions of lysosomes would be indispensable for neurons to clear misfolded proteins and damaged organelles, and their movement is crucial to many cellular functions, while anterograde axonal transport is kinesin-mediated and retrograde transport depends on the dynein motor [Citation57]. Importantly, TMT impaired lysosomal anterograde transport in primary hippocampal neurons, and reduced the levels of KIF5A but not DYNLL1 (dynein light chain LC8-type 1) or DCTN1 (dynactin subunit 1) (Table S5 and Figure S13). Overexpression of Kif5a significantly rescued lysosomal anterograde transport. Therefore, KIF5A reduction-dependent axonal transport defects may be involved in TMT-induced lysosomal dysfunction and autophagic flux impairment.
TMT can impair the hippocampus of both humans and animals since it is soluble in both water and lipids and can easily cross the blood-brain barrier [Citation8]; it induces two typical phenomena in the acute phase: histomorphological injury of the hippocampus and seizures [Citation58,Citation59]. In rodents, TMT induces an extensive neuronal loss in the hippocampus characterized by swelling, nuclear pyknosis, karyolysis, and necrosis [Citation8,Citation60]. These injuries are closely associated with behavioral abnormalities, including ataxia, aggressiveness, tail mutilation, vocalization, and seizures [Citation61,Citation62]. Among them (these symptoms), a seizure is the most representative symptom in TMT-administered rodents [Citation63]. Consistent with previous studies, we observed remarkable histomorphological injury of the hippocampus and seizures in vivo after TMT administration. Notably, KIF5A deficiency induces epilepsy in both humans [Citation64] and mice [Citation65] and is associated with myoclonic seizures in humans [Citation66]. In our study, TMT obviously decreased KIF5A expression, and the overexpression of Kif5a effectively alleviated TMT-induced seizures, suppressed histomorphological injury of the hippocampus, and restored autophagy flux. Accordingly, our results suggested that the activation of KIF5A-dependent autophagy contributes to the antagonism of TMT-induced neurotoxicity in vivo.
Collectively, these results provide new evidence of KIF5A-dependent axonal transport deficiency, causing dysfunctional autophagy in TMT-induced neurotoxicity (). The new findings of the present study bridge the knowledge gap between autophagy and TMT-induced neurotoxicity. They may also have implications for understanding the roles of axonal transport and autophagy in the development of neurological disorders induced by environmental stress.
Materials and methods
Cell culture and TMT treatment
Mouse neuroblastoma cells (Neuro-2a cells) were obtained from the Cell Bank of the Institute of Biochemistry, and Cell Biology (Shanghai, China, TCM29) and cultured with DMEM/H (HyClone, SH30022.01) supplemented with 10% fetal bovine serum (FBS; PAN-Biotech, P30-3302) and 1% (v:v) penicillin/streptomycin (Sigma-Aldrich, P4333). The cells were grown in a 5% CO2 humidified atmosphere at 37°C. TMT (Sigma-Aldrich, 146,498) and CQ (Sigma-Aldrich, C6628) were dissolved in distilled deionized water to produce a stock solution, which was then appropriately diluted with cell culture medium before application. 3-MA (Sigma-Aldrich, M9281), a poorly water-soluble drug, was dissolved with sterilized phosphate-buffered saline (PBS; Beyotime, C0221A) solution to produce a stock solution of 100 mM 3-MA. Before each trial, the stock solution was heated in a water bath at 50°C and then diluted into working concentration with culture medium, avoiding white flock formation [Citation67]. For TMT treatment, Neuro-2a cells were grown to 70 ~ 80% confluence and treated with different concentrations (0, 2, 4, 8 μM) of TMT for 24 h [Citation18]. For CQ treatment, Neuro-2a cells were treated with 8 μM TMT with or without CQ (50 μM) for 24 h [Citation68]. For 3-MA treatment, Neuro-2a cells were treated with 8 μM TMT with or without 3-MA (2 mM) for 24 h [Citation14].
Primary hippocampal neurons were isolated from C57BL/6 newborn mice (P0-P1) under stereomicroscope [Citation69,Citation70]. Dissociated hippocampal neurons were cultured in poly-L-lysine (Sigma-Aldrich, P4832)-coated glass bottom dishes with DMEM/F-12 medium (Gibco, 11,330,032) containing 10% FBS, 10% horse serum (Gibco, 26,050,088) and 1% (v:v) penicillin/streptomycin in a 5% CO2 humidified atmosphere at 37°C. At 24 h after seeding, the medium was replaced by Neurobasal-A Medium (Gibco, 10,888,022) supplemented with 2% B-27 (Gibco, A3582801) and 1% GlutaMAX (Gibco, 35,050,061) for 7 d for the assay. Primary hippocampal neurons were treated with 0, 0.5, 1 and 2 μM TMT for 24 h [Citation71].
Preparation of AAV-Kif5a
To overexpress Kif5a in the CNS in vivo, the AAV PHP.eB was generated from OBio Technology (Shanghai, China) and used as a vector [Citation72]. Kif5a cDNA was acquired by digesting a Kif5a plasmid (Sangon Biotech, China) and extracting the DNA fragment from an agarose gel using the MiniBEST Agarose Gel DNA Extraction Kit Ver. 3.0 (Takara, 9762). Kif5a cDNA was subsequently packaged it into an AAV PHP.eB vector (pAAV-CMV-Kif5a-3FLAG -CW3SL, AAV-Kif5a; titer: 6.27 × 1012 V.G./mL, 5.47 × 1012 V.G./mL and 9.8 × 1012 V.G./mL) by OBio Technology. The control was an empty vector (AAV-null; titer: 1.52 × 1013 V.G./mL and 1.7 × 1013 V.G./mL).
Animal administration and experimental design
Male C57BL/6 J mice were purchased from Vital River Laboratories (Beijing, China) and housed two to three per cage with ad libitum access to food and water with a 12-h light/dark cycle. All animal experimental procedures were conducted in accordance with institutional animal welfare guidelines and were approved by the Third Military Medical University Animal Care and Use Committee.
To investigate TMT-induced autophagic flux dysfunction and neurotoxicity in vivo, 8-week-old C57BL/6 J mice were randomly divided into 2 groups: (1) the control group mice (n = 25/group) and (2) the TMT group mice (n = 25/group). The TMT group received a single intraperitoneal (i.p.) injection of 2.8 mg/kg TMT dissolved in saline. The control group mice received an injection of the same volume of saline [Citation73]. After TMT treatment for 24 h, the mice were perfused transcardially with 0.9% saline under pentobarbital anesthesia. Hippocampal tissues were collected from the sacrificed mice and placed in liquid nitrogen. The samples were stored at −80°C until use.
To investigate the protective effect of KIF5A on TMT-induced autophagic flux dysfunction and neurotoxicity in vivo, 5-week-old C57BL/6 J mice were randomly and equally divided into three groups: (1) the AAV-null+control group (n = 25/group), (2) the AAV-null+TMT group (n = 25/group) and (3) the AAV-Kif5a+TMT group (n = 25/group). For the AAV-Kif5a+TMT group, the mice were intravenously injected with 2 × 1011 AAV-Kif5a dissolved in saline. For all AAV-null groups, the mice received the same dose of AAV-null by injection. After 3 weeks, the AAV-null+TMT and AAV-Kif5a+TMT group mice were administered (i.p.) 2.8 mg/kg TMT, and the AAV-null+control group mice received the same volume of saline by injection. After TMT treatment for 24 h, the mice were perfused transcardially with 0.9% saline under pentobarbital anesthesia. Hippocampal tissues were collected from the sacrificed mice and placed in liquid nitrogen. The samples were stored at −80°C until use.
To investigate the efficiency of Kif5a overexpression in C57BL/6 J mice, 5-week-old C57BL/6 J mice were randomly and equally divided into two groups: (1) the AAV-null group (n = 10/group), (2) the AAV-Kif5a group (n = 10/group). For the AAV-Kif5a group, the mice were intravenously injected with 2 × 1011 AAV-Kif5a dissolved in saline, and the AAV-null group received the same dose of AAV-null. After 3 weeks, the mice were perfused transcardially with 0.9% saline under pentobarbital anesthesia. Hippocampal tissues were collected from the sacrificed mice and placed in liquid nitrogen. The samples were stored at −80°C until use.
Adenovirus upregulation of Kif5a
An adenovirus expressing mouse Kif5a gene, Ad/Kif5a, and adenovirus expressing a control (null) gene, Ad/control, were both commercially purchased from OBio Technology (Shanghai, China). Two days prior to TMT treatment, primary hippocampal neurons were infected with Ad/control or Ad/Kif5a for 48 h.
Cell viability assay
The Cell Counting Kit-8 (CCK-8; Dojindo, CK04) assay was used to measure cell viability according to the manufacturer`s instructions [Citation74]. Neuro-2a cells (1 × 104) were seeded in 96-well plates, and primary hippocampal neurons (2 × 104) were seeded in poly-L-lysine-coated 96-well plates Then, 100 μL of medium containing 10% CCK-8 reagent was added to a subset of wells after different treatments and incubated for 45 min at 37°C. Then, the OD value was measured at 450 nm using an Infinite™ M200 Microplate Reader (Tecan, Switzerland).
Cell morphological observation
For morphological observation, 1 × 104 cells were seeded in 96-well plates at 70 ~ 80% confluence and treated with different concentrations (0, 2, 4, 8 μM) of TMT for 24 h. Then, the cells were observed using an inverted microscope (LEICA DM IRB, Germany).
Tandem mass tag-based quantitative proteomic analysis
Tandem mass tag-based quantitative proteomic analysis was performed by Applied Protein Technology (Shanghai, China). The treated cells were put into SDT lysis buffer (4% SDS, 100 mM Tris-HCl, 1 mM DTT, pH 7.6) and then boiled for 15 min. After centrifugation at 14,000 × g for 40 min, the supernatant was quantified with the BCA Protein Assay Kit (Beyotime, P0012). Two hundred micrograms of proteins for each sample were digested by the filter-aided sample preparation method [Citation75], and the peptide content was estimated by UV light spectral density at 280 nm. Then, 100 µg of the resulting peptide mixture was labeled using Tandem Mass Tag™ Isobaric Mass Tagging Kits and Reagents (Thermo Scientific, 90113CH) and fractionated using the Pierce High pH Reversed-Phase Peptide Fractionation Kit (Thermo Scientific, 84,868) according to the manufacturer’s instructions. Each fraction was injected for nanoLC-MS/MS analysis. The peptide mixture was loaded onto a reversed-phase trap column (Acclaim PepMap100, 100 μm*2 cm, nanoViper C18, 3 μm, 100 Å; Thermo Scientific, USA) connected to a C18-reverse-phase analytical column (Easy Column, 10 cm long, 75 μm inner diameter, 3 μm resin, C18-A2; Thermo Scientific, USA) in buffer A (0.1% formic acid; Fluka, 06450) and separated with a linear gradient of buffer B (84% acetonitrile [Merck, I592230123] and 0.1% formic acid) at a flow rate of 300 nL/min controlled by IntelliFlow technology (Aptbio, Shanghai, China). LC-MS/MS analysis was performed on a Q Exactive mass spectrometer (Thermo Scientific, USA) that was coupled to an Easy-nLC 1000 instrument (Thermo Scientific, USA) for 90 min. The mass spectrometer was operated in positive ion mode. MS data were acquired using a data-dependent top10 method, dynamically choosing the most abundant precursor ions from the survey scan (300–1800 m/z) for HCD fragmentation. The automatic gain control (AGC) target was set to 3e6, and the maximum inject time was set to 10 ms. The dynamic exclusion duration was 40.0 s. Survey scans were acquired at a resolution of 70,000 at 200 m/z, and the resolution for the HCD spectra was set to 17,500 at 200 m/z (Tandem Mass Tag 6plex), and 35,000 at 200 m/z (Tandem Mass Tag 10plex), and the isolation width was 2 m/z. The normalized collision energy was 30 eV, and the underfill ratio, which specifies the minimum percentage of the target value likely to be reached at maximum fill time, was defined as 0.1%. The instrument was run with peptide recognition mode enabled. MS/MS spectra were searched using MASCOT engine (version 2.2; Matrix Science, UK) embedded into Proteome Discoverer 1.4 (Thermo Scientific, USA).
Bioinformatics analysis
To investigate the role of diverse pathways in TMT neurotoxicity, pathway analysis was performed using the Kyoto Encyclopedia of Genes and Genomes database (https://www.genome.jp/kegg/pathway.html) and Ingenuity Pathway Analysis (https://www.qiagenbioinformatics.com/; QIAGEN, USA). To investigate possible molecular mechanisms, IPA was conducted to analyze the molecular interaction network among the proteins identified by LC-MS/MS.
Western blot analysis
The cells or tissue samples of 8 ~ 9 mice were lysed in RIPA Lysis Buffer (Beyotime, P0013B) with cOmplete™, Mini, EDTA-free Protease Inhibitor Cocktail (Roche, 04693124001). After incubation on ice for 30 min, the protein concentrations of the supernatants of the lysates collected after centrifugation for 15 min at 12,000 × g were measured using a BCA kit (Beyotime, P0011). The total protein was boiled in SDS loading buffer (Beyotime, P0015) and separated using 8-15% sodium dodecyl sulfate-polyacrylamide gel electrophoresis. Subsequently, the proteins were transferred to polyvinylidene fluoride membranes (Bio-Rad, 1,704,272), blocked in 5% nonfat dry milk (Boster, AR0104) for 1 h at room temperature, and then incubated overnight at 4°C with antibodies against LC3B (Sigma-Aldrich, L7543), SQSTM1 (Abcam, ab56416), LAMP1 (Invitrogen, MA1-164), LAMP2 (Invitrogen, MA1-205 and PA1-655), KIF5A (Abcam, ab5628), ATP6V1E1 (Bioworld Technology, BS72008), ATP6V0D1 (Bioworld Technology, BS5977), ATG5 (Cell Signaling Technology, 12,994S), DYNLL1 (Invitrogen, MA1-070), DCTN1 (Invitrogen, PA5-18,095), ACTB (actin beta; Sigma-Aldrich, A1978), TUB (tubulin; Beyotime, AT819) and GAPDH (glyceraldehyde-3-phosphate dehydrogenase; Abcam, ab8245). The membranes were washed three times in 1 × TBST (Sangon Biotech, C520009) and then incubated with the corresponding HRP-conjugated anti-mouse, anti-rabbit or anti-goat (Beyotime, A0208, A0216 or A0181) secondary antibodies for 1 h at room temperature. Next, the membranes were washed and visualized using an Immobilon Forte Western HRP substrate (Millipore, WBLUF0500). The bands were quantified with ImageJ software (NIH). The relative optical density of the target protein of each group was calculated compared to the optical density of the ACTB, TUB, or GAPDH proteins [Citation76].
Transfection
Based on the manufacturer’s instructions [Citation77], Neuro-2a cells were transfected with Opti-MEM® I reduced serum media (Gibco, 31,985,062) and Lipofectamine™ 2000 Transfection Reagent (Invitrogen, 11,668–019). The cells were transfected with 2 μg/mL GFP-LC3B plasmid (Cell Biolabs, CBA-401) or 0.8 μg/mL Kif5a plasmid (Sangon Biotech, China), together with the same concentration of a pcDNA 3.0 plasmid (Sangon Biotech, China) as a control plasmid. Atg5 siRNA (100 nmol/L; Santa Cruz Biotechnology, sc-41,446) or a control siRNA (100 nmol/L; Santa Cruz Biotechnology, sc-37,007) were used for siRNA-mediated silencing. After transfection for 24 h, the cells were used for subsequent experiments.
Immunofluorescence analysis of Neuro-2a cells
Immunofluorescence was performed according to standard procedures [Citation78]. In brief, cells were grown on gelatin-coated glass coverslips. After pre-stage processing, the cells were fixed with 4% paraformaldehyde (PFA; Beyotime, P0099) in PBS for 30 min, followed by permeabilization with 0.25% Triton X-100 (Sigma-Aldrich, T8787) in PBS for 10 min at room temperature. Then, the cells were blocked with 10% BSA in PBS. The fixed cells were incubated with mouse anti-SQSTM1 (1:100; Abcam, ab56416), rat anti-LAMP2 (1:100; Abcam, ab13524) or rabbit anti-KIF5A (1:200; Abcam, ab5628) in immunostaining dilution buffer at 4°C overnight. The slides were then washed 5 times with PBS and incubated with an Alexa Fluor® 568 donkey anti-rabbit IgG (H + L) antibody (Invitrogen, A10042), an Alexa Fluor® 647 goat anti-rat IgG (H + L) antibody (Invitrogen, A21247) or an Alexa Fluor® 568 donkey anti-mouse IgG (H + L) antibody (Invitrogen, A10037) based on the source of the primary antibody, at 1:200 dilutions for 1 h at 37°C. DAPI Staining Solution (Beyotime, C1005) was used for nuclear counterstaining. The coverslips were mounted on the glass slides using antifade mounting medium (Beyotime, P0126). The stained samples were examined using a ZEISS LSM800 confocal laser scanning microscope (ZEISS, Germany) equipped with a 63 × or 40 × oil objective. The colocalization coefficient was calculated using ImageJ software. At least 30 cells were counted for each experiment.
RFP-GFP-LC3B assay
Premo™ Autophagy Tandem Sensor RFP-GFP-LC3B (Invitrogen, P36239), which is a lentivirus carrying expression cassettes that encode tandem fluorescence-tagged LC3B, was used to evaluate the number of autophagosomes and autolysosomes [Citation14,Citation20]. Briefly, 2 × 105 Neuro-2a cells were grown on glass-bottom dishes and infected with lentivirus for 24 h. Then, the Neuro-2a cells were treated with TMT for an additional 24 h. All samples were examined under a ZEISS LSM 780 confocal laser scanning microscope (ZEISS, Germany) equipped with a 40 × or 63 × oil immersion objective.
DQ-BSA proteolytic activity assay
To examine lysosomal proteolysis activity, 1 × 104 cells were plated in 96-well plates. Upon 80% confluency, the cells were first loaded with 10 μg/mL DQ™ Red BSA (Life Technologies, D-12,051) at 37°C for 6 h. Then, the cells were washed 3 times in PBS, followed by treatment with other reagents. The fluorescence intensity of the cells was quantified using an Infinite™ M200 Microplate Reader with 590-nm excitation and 620-nm emission filters.
LysoSensor Green DND-189 staining
The quantification of the lysosomal pH was performed using LysoSensor Green DND-189 (Invitrogen, L7535). Briefly, the cells were loaded with 1 μM LysoSensor Green DND-189 in prewarmed regular medium for 5 min at 37°C. Then, the cells were washed twice with PBS. After treatment, the fluorescence intensity of the cells was quantified using an Infinite™ M200 Microplate Reader with 485-nm excitation and 530-nm emission filters.
Live-cell imaging
Microfluidic chambers (Xona Microfluidics LLC, SND450) with microgroove width of 450 μm allows axons to extend to the adjacent compartment but excludes dendrites [Citation29]. Microfluidic chambers were cleaned with a 1% Alconox solution (Sigma-Aldrich, Z273228) in MilliQ water, rinsed 6 times with MilliQ water, dipped in 70% ethanol, and left to dry in cell culture hood for 20 min under ultraviolet light. 5 × 104 primary hippocampal neurons were plated in poly-L-lysine-coated microfluidic chambers. A higher volume of media was maintained in the axonal compartment, and from this time until use, a higher volume of media was maintained in the soma compartment [Citation79]. After different treatment, primary hippocampal neurons incubated in medium containing 100 nM LysoTracker™ Red DND-99 (Invitrogen, L7528) for 30 min at 37°C. Cells were washed and then replaced with fresh culture medium before imaging. Live-cell imaging was performed using a ZEISS LSM800 confocal laser scanning microscope (ZEISS, Germany) equipped with a 40 × oil objective and an Incubator PM 2000 RBT (PeCon, Germany) set at 37°C and 5% CO2. Time-lapse images were captured at 500 ms/frame for 2 min and processed and analyzed using ImageJ. Kymographs were generated using ImageJ plugin-KymoToolBox (https://github.com/fabricecordelieres/IJ_KymoToolBox). Anterograde and retrograde motile vesicles were defined as particles showing a net displacement of ≥ 3 μm in one direction [Citation80–82].
PRM MS analysis
About 200 μg protein of each sample added DTT to a final concentration of 100 mM. The samples were boiled for 15 min and cooled to room temperature. Then, each sample was mixed with 200 μL UA buffer (8 M urea [Bio-Rad, 161–0731], 150 mM Tris-HCl, pH 8.0), and centrifugated in ultrafiltration tube at 14,000 × g for 40 min. Discard the filtrate and add 100 μL IAA buffer (50 mM IAA [Bio-Rad, 163–2109] in UA) to shake at 150 × g for 1 min. After incubation at room temperature for 30 min, each sample was centrifuged at 14,000 × g for 20 min. Subsequently, each sample was washed with 100 μL UA buffer 3 times and 100 μL NH4CO3 buffer (50 mM) for 2 times at 14,000 × g for 20 min. After adding 40 μL NH4CO3 buffer containing trypsin (1:50; Promega, 317,107), the samples were shaken at 150 × g for 1 min and incubated for 16 h at 37°C. Centrifugation at 14,000 × g for 20 min and another 30 min following adding 40 μL NH4HCO3 buffer was used to collect the peptides. After desalting and lyophilization, the peptides were redissolved in buffer A. The peptide content was estimated by UV light spectral density at 280 nm. The peptide mixture was loaded onto a home-made trap column (100 μm*50 mm, 5 μm-C1) connected to a home-made tip column (75 μm*200 mm, 3 μm-C18) in buffer A and separated at 300 nL/min for 2 min from 5 to 10% buffer B (98% acetonitrile, 0.1% formic acid), a 43 min linear gradient to from 10 to 30% buffer B and a 10 min linear gradient to from 30 to 100% buffer B, maintained at 100% buffer B for 5 min. LC-PRM/MS analysis (Aptbio, Shanghai, China) was performed by Q-Exactive HF mass spectrometer (Thermo Scientific, USA) that was coupled to an Easy-nLC 1000 instrument (Thermo Scientific, USA) for 60 min. The mass spectrometer was operated in positive ion mode. The full MS scan was taken at a resolution of 60,000 at 200 m/z with a scan mass range of 350 to 1800 m/z, a target AGC of 3e6, and maximum inject time is 200 ms. After each full MS scan, 20 PRM scans (MS2 scans) were collected based on Inclusion list at an isolation window of 1.6 Th, a resolution of 30,000 at 200 m/z, a target AGC of 3e6, and maximum injection fill time is 120 ms. Peptides were selected for MS2 scans using HCD operating mode with a normalized collision energy setting of 27%. The sum of the top three fragment ion intensities was calculated in Skyline (MacCoss Lab Software version 3.5.0) and used to estimate peptide signal intensity. Peptide concentration was calculated based on the ratio to the heavy peptide standards that were added in a known quantity.
Assessment of seizure severity
To assess the typical tremor/seizure behavior of the mice, a rating scale, as described by Jingang Hou et al., was used [Citation58]. Twenty-four hours after TMT injection, twenty mice per group were scored during a 5-min interval on a tremor/seizure severity scale as follows: 1, normal behavior; 2, hyper-responsiveness to sound and handling; 3, whole-body mild tremor with normal motor activity; 4, whole-body tremor with extended periods of immobility; 5, rigid posture; 6, forelimb clonus, rearing and falling; 7, repeated incidence of level 4 behavior; and 8, severe tonic-clonic behavior.
Nissl staining
Four treated animals per group were perfused transcardially with 0.9% saline, followed by 4% PFA in PBS under pentobarbital anesthesia. The removed brains were fixed overnight in 4% PFA. Then, the brain tissues were dehydrated, cleared, embedded in paraffin, and cut into 4-μm thick coronal sections. The sections were mounted on slides and baked in an oven at 65°C for 1 h. For Nissl staining, the sections were stained with 0.5% toluidine blue (Sigma-Aldrich, T3260) for 10 min. All the sections were coverslipped with resinene, and digital images were acquired with an ECLIPSE Ci-S microscope (NIKON, Japan). Compared with the normal cells with regular shape and even staining, the injured neuronal cells in the hippocampus were loss, nuclear shrinkage and stained darkly. For each mouse, one of the representative coronal brain sections was selected for statistical evaluation. The number of the injured cells at 200× magnification within the hippocampus DG, CA1, and CA3 regions were counted [Citation83,Citation84].
Immunofluorescence analysis of mouse brain sections
Four treated animals per group were perfused transcardially with 0.9% saline followed by 4% PFA in PBS under pentobarbital anesthesia. The removed brains were fixed overnight in 4% PFA and dehydrated in 30% sucrose. The dehydrated brains were embedded in OCT at −20°C and sectioned at a thickness of 20 μm. The sections were washed three times with 0.3% Triton X-100/PBS solution for 10 min and blocked in 3% BSA and 0.3% Triton X-100 in PBS for 1 h at room temperature. The sections were incubated in primary antibody solutions overnight at 4°C. The primary antibodies used included rabbit anti-LC3B (1:200; Sigma-Aldrich, L7543) and rabbit anti-KIF5A (1:200; Abcam, ab5628). The sections were further incubated with the appropriate Alexa Fluor® 568 donkey anti-rabbit IgG (H + L) antibody for 2 h at room temperature. The cell nuclei were visualized by staining with DAPI staining solution (Beyotime, C1005) for 10 min at room temperature. Then, the sections were mounted on glass slides, and digital images were acquired at 200 × magnification using a ZEISS LSM800 confocal laser scanning microscope (ZEISS, Germany) [Citation14].
CTSD activity assay
The Cathepsin D Activity Fluorometric Assay Kit (BioVision, K143-100) was used to detect CTSD catalytic activity in the hippocampi of 9 ~ 10 mice. Briefly, the hippocampus was removed from the hemicerebrum and ground in 200 μL of chilled CD Cell Lysis Buffer for 60 s at 30 Hz with a TissueLyser II (QIAGEN, 85,300), followed by a 10-min incubation on ice. After centrifugation, 50 μL of the supernatant was transferred to a 96-well plate, and 52 μL of master assay mix containing 50 μL of CD reaction buffer and 2 μL of 10 mM CD substrate was added to each sample. The samples were incubated at 37°C for 30 min and were read with an Infinite™ M200 Microplate Reader with 328-nm excitation and 460-nm emission filters. The activity was normalized to the protein concentrations of the samples [Citation85].
CTSB activity assay
The Cathepsin B Activity Fluorometric Assay Kit (BioVision, K140-100) was used to detect CTSB catalytic activity in the hippocampi of 9 ~ 10 mice. Briefly, the hippocampus was removed from the hemicerebrum and ground in 200 μL of chilled CB cell lysis buffer with a TissueLyser II, followed by a 10-min incubation on ice. After centrifugation, 50 μL of the supernatant was transferred to a 96-well plate, and 50 μL of CB reaction buffer and 2 μL of 10 mM CB substrate Ac-RR-AFC was added to each sample. The samples were incubated at 37°C for 30 min and were read with an Infinite™ M200 Microplate Reader with 400-nm excitation and 505-nm emission filters. The activity was normalized to the protein concentrations of the samples [Citation86].
Measurement of intralysosomal pH in the hippocampus
The measurement of intralysosomal pH in the hippocampus was performed according to the previous study [Citation87]. Briefly, hippocampi were removed from the hemicerebrum and trypsinized by papain (Roche, 10,108,014,001) at 37°C. Then, the tissues were triturated with DMEM/F-12 medium containing 10% FBS and 10% horse serum, filtered (70 μm), and centrifuged at 155 g for 5 min at 4°C. The pellet was resuspended and incubated with 1 μM LysoSensor Green DND-189 in the medium for 5 min at 37°C. After washing with ice-cold PBS, samples were detected by flow cytometry (Accuri C6; BD Bioscience, USA).
Total RNA extraction and quantitative real-time PCR analysis
The hippocampi from the cerebral hemisphere of 8 mice were ground in RNAiso Plus (TaKaRa, 9109) for 60 s at 30 Hz with a TissueLyser II. The total RNA was isolated from the lysates, and the concentration was determined using a NanoDrop 2000 spectrophotometer (Thermo Scientific, USA). One microgram of total RNA was used for cDNA synthesis using the PrimeScript™ RT Reagent Kit with gDNA Eraser (Takara, RR047A). TB Green™ Premix Ex Taq™ II (TaKaRa, RR820A) was used as a fluorescent dye, and all the primers used in this experiment are shown in Table S3. Quantitative real-time PCR was performed using the iQ5 Real-Time PCR Detection System (Bio-Rad, USA). The mean fold-change is shown as the natural logarithm of the RQ values, and the error was estimated by evaluating the 2−ΔΔCt equation using ΔΔCt plus the standard deviation and ΔΔCt minus the standard deviation [Citation88].
Immunoprecipitation
The cells were lysed in Cell Lysis Buffer for Western and IP (Beyotime, P0013) with cOmplete™, Mini, EDTA-free Protease Inhibitor Cocktail. The lysates were clarified by centrifugation at 12,000 × g for 15 min and incubated with 2 μg of an antibody against LAMP2 (Abcam, ab13524) overnight at 4°C. Then, Protein A Agarose (Beyotime, P2006) was added to the lysates and incubated at 4°C for 4 h. After 3 washes with PBS, the precipitates were solubilized with 20 μL of lysis buffer and 20 μL of SDS loading buffer (Beyotime, P0015). Subsequently, the samples were denatured and analyzed by immunoblotting with the antibodies against LAMP2 (Invitrogen, MA1-205) and KIF5A (Abcam, ab5628). Secondary antibodies were used anti-mouse IRDye 800CW (LI-COR Biosciences, 926–33,210) and anti-rabbit IRDye 680RD (LI-COR Biosciences, 926–68,071). The membranes were visualized using the Odyssey Imaging System [Citation89].
Statistical analysis
The data are expressed as the mean ± SEM. Comparisons of the data among the groups were performed using one-way ANOVA (Bonferroni’s multiple-comparison test) for parametric (normality and equal variance passed) data. Kruskal-Wallis ANOVA based on ranks followed by Dunn’s posthoc test was used for nonparametric (normality and/or equal variance failed) data. For experiments with only two groups, a two-tailed Mann-Whitney rank-sum test (nonparametric) or a two-tailed unpaired Student’s t-test was performed. In all analysis, the null hypothesis was rejected at the 0.05 level. All the counting analysis were performed in a blinded manner, and colocalization was confirmed using 3D images.
Supplemental Material
Download Zip (115.4 MB)Acknowledgments
We’d like to show our great appreciation to Shanghai Applied Protein Technology Co., Ltd. for technical support of proteomics.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary Material
Supplemental data for this article can be accessed here.
Additional information
Funding
References
- Boyer IJ. Toxicity of dibutyltin, tributyltin and other organotin compounds to humans and to experimental animals. Toxicology. 1989 May 15;55(3):253–298. PubMed PMID: 2655175.
- Feldman RG, White RF, Eriator II. Trimethyltin encephalopathy. Arch Neurol. 1993 Dec;50(12):1320–1324. PubMed PMID: 8257310.
- Yoo CI, Kim Y, Jeong KS, et al. A case of acute organotin poisoning. J Occup Health. 2007 Jul;49(4):305–310. PubMed PMID: 17690524.
- Guo F, Lu XW, Xu QP. Diagnosis and treatment of organotin poisoned patients. World J Emerg Med. 2010;1(2):122–125. PubMed PMID: 25214954; PubMed Central PMCID: PMCPMC4129753.
- Xiao L, Zou K. Electromyography analysis in 13 patients with acute trimethyltin chloride poisoning. Zhonghua Lao Dong Wei Sheng Zhi Ye Bing Za Zhi. 2018 Sep 20;36(9):685–687. PubMed PMID: 30419673.
- Lee S, Yang M, Kim J, et al. Trimethyltin-induced hippocampal neurodegeneration: A mechanism-based review. Brain Res Bull. 2016 Jul;125:187–199. PubMed PMID: 27450702.
- Trabucco A, Di Pietro P, Nori SL, et al. Methylated tin toxicity a reappraisal using rodents models. Arch Ital Biol. 2009 Dec;147(4):141–153. PubMed PMID: 20162863.
- Tang X, Wu X, Dubois AM, et al. Toxicity of trimethyltin and dimethyltin in rats and mice. Bull Environ Contam Toxicol. 2013 May;90(5):626–633. PubMed PMID: 23478947.
- Zhang L, Zhao Q, Chen CH, et al. Synaptophysin and the dopaminergic system in hippocampus are involved in the protective effect of rutin against trimethyltin-induced learning and memory impairment. Nutr Neurosci. 2014 Sep;17(5):222–229. PubMed PMID: 24001577.
- Galluzzi L, Bravo-San Pedro JM, Blomgren K, et al. Autophagy in acute brain injury. Nat Rev Neurosci. 2016 Aug;17(8):467–484. PubMed PMID: 27256553.
- Hansen M, Rubinsztein DC, Walker DW. Autophagy as a promoter of longevity: insights from model organisms. Nat Rev Mol Cell Biol. 2018 Sep;19(9):579–593. PubMed PMID: 30006559.
- Lumkwana D, Du Toit A, Kinnear C, et al. Autophagic flux control in neurodegeneration: progress and precision targeting-where do we stand? Prog Neurobiol. 2017 Jun;153:64–85. PubMed PMID: 28385648.
- Pellacani C, Costa LG. Role of autophagy in environmental neurotoxicity. Environ Pollut. 2018 Apr;235:791–805. PubMed PMID: 29353798.
- Li M, Pi H, Yang Z, et al. Melatonin antagonizes cadmium-induced neurotoxicity by activating the transcription factor EB-dependent autophagy-lysosome machinery in mouse neuroblastoma cells. J Pineal Res. 2016 Oct;61(3):353–369. PubMed PMID: 27396692.
- Fabrizi C, Somma F, Pompili E, et al. Role of autophagy inhibitors and inducers in modulating the toxicity of trimethyltin in neuronal cell cultures. J Neural Transm (Vienna). 2012 Nov;119(11):1295–1305. PubMed PMID: 22415064.
- Fabrizi C, Pompili E, De Vito S, et al. Impairment of the autophagic flux in astrocytes intoxicated by trimethyltin. Neurotoxicology. 2016 Jan;52:12–22. PubMed PMID: 26459185.
- Fabrizi C, Pompili E, Somma F, et al. Lithium limits trimethyltin-induced cytotoxicity and proinflammatory response in microglia without affecting the concurrent autophagy impairment. J Appl Toxicol. 2017 Feb;37(2):207–213. PubMed PMID: 27226005.
- Zhao W, Pan X, Li T, et al. Lycium barbarum polysaccharides protect against trimethyltin chloride-induced apoptosis via sonic hedgehog and PI3K/Akt signaling pathways in mouse neuro-2a cells. Oxid Med Cell Longev. 2016;2016:9826726. PubMed PMID: 27143997; PubMed Central PMCID: PMCPMC4838808.
- Schaaf MB, Keulers TG, Vooijs MA, et al. LC3/GABARAP family proteins: autophagy-(un)related functions. Faseb J. 2016 Dec;30(12):3961–3978. PubMed PMID: 27601442.
- Klionsky DJ, Abdelmohsen K, Abe A, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy. 2016;12(1):1–222. PubMed PMID: 26799652; PubMed Central PMCID: PMCPMC4835977.
- Svenning S, Johansen T. Selective autophagy. Essays Biochem. 2013;55:79–92. PubMed PMID: 24070473.
- Mauthe M, Orhon I, Rocchi C, et al. Chloroquine inhibits autophagic flux by decreasing autophagosome-lysosome fusion. Autophagy. 2018;14(8):1435–1455. PubMed PMID: 29940786; PubMed Central PMCID: PMCPMC6103682.
- Ganley Ian G, Wong P-M, Gammoh N, et al. Distinct autophagosomal-lysosomal fusion mechanism revealed by thapsigargin-induced autophagy arrest. Mol Cell. 2011;42(6):731–743.
- Kirkin V, McEwan DG, Novak I, et al. A role for ubiquitin in selective autophagy. Mol Cell. 2009 May 15;34(3):259–269. PubMed PMID: 19450525.
- Manley S, Ni HM, Kong B, et al. Suppression of autophagic flux by bile acids in hepatocytes. Toxicol Sci. 2014 Feb;137(2):478–490. PubMed PMID: 24189133; PubMed Central PMCID: PMCPMC3908720.
- Lu Y, Dong S, Hao B, et al. Vacuolin-1 potently and reversibly inhibits autophagosome-lysosome fusion by activating RAB5A. Autophagy. 2014;10(11):1895–1905. PubMed PMID: 25483964; PubMed Central PMCID: PMCPMC4502727.
- Morfini G, Schmidt N, Weissmann C, et al. Conventional kinesin: biochemical heterogeneity and functional implications in health and disease. Brain Res Bull. 2016 Sep;126(Pt 3):347–353. PubMed PMID: 27339812.
- Pu J, Guardia CM, Keren-Kaplan T, et al. Mechanisms and functions of lysosome positioning. J Cell Sci. 2016 Dec 1;129(23):4329–4339. PubMed PMID: 27799357; PubMed Central PMCID: PMCPMC5201012.
- Taylor AM, Blurton-Jones M, Rhee SW, et al. A microfluidic culture platform for CNS axonal injury, regeneration and transport. Nat Methods. 2005 Aug;2(8):599–605. PubMed PMID: 16094385; PubMed Central PMCID: PMCPMC1558906.
- Ooigawa H, Nawashiro H, Fukui S, et al. The fate of Nissl-stained dark neurons following traumatic brain injury in rats: difference between neocortex and hippocampus regarding survival rate. Acta Neuropathol. 2006 Oct;112(4):471–481. PubMed PMID: 16858608.
- Wassmer T, Sehring IM, Kissmehl R, et al. The V-ATPase in paramecium: functional specialization by multiple gene isoforms. Pflugers Arch. 2009 Jan;457(3):599–607. PubMed PMID: 18228038.
- Tang X, Li N, Kang L, et al. Chronic low level trimethyltin exposure and the risk of developing nephrolithiasis. Occup Environ Med. 2013 Aug;70(8):561–567. PubMed PMID: 23703823.
- Zhu HB, Ouyang GL, Lai YY, et al. Characteristics of clinical, magnetic resonance imaging and electroencephalogram after trimethyltin chloride poisoning. Zhonghua Lao Dong Wei Sheng Zhi Ye Bing Za Zhi. 2019 Feb 20;37(2):133–136. PubMed PMID: 30929356.
- Wang M, Li B, Wang C, et al. The concentration-dependent induction of cell death by trimethyltin chloride in rat liver epithelial IAR20 cells. Toxicol In Vitro. 2008 Aug;22(5):1136–1142. PubMed PMID: 18411021.
- Ross WD, Emmett EA, Steiner J, et al. Neurotoxic effects of occupational exposure to organotins. Am J Psychiatry. 1981 Aug;138(8):1092–1095. PubMed PMID: 7258389.
- Kreyberg S, Torvik A, Bjorneboe A, et al. Trimethyltin poisoning: report of a case with postmortem examination. Clin Neuropathol. 1992 Sep-Oct;11(5):256–259. PubMed PMID: 1424320.
- Angel TE, Aryal UK, Hengel SM, et al. Mass spectrometry-based proteomics: existing capabilities and future directions. Chem Soc Rev. 2012 May 21;41(10):3912–3928. PubMed PMID: 22498958; PubMed Central PMCID: PMCPMC3375054.
- Larance M, Lamond AI. Multidimensional proteomics for cell biology. Nat Rev Mol Cell Biol. 2015 May;16(5):269–280. PubMed PMID: 25857810.
- Lattanzi W, Corvino V, Di Maria V, et al. Gene expression profiling as a tool to investigate the molecular machinery activated during hippocampal neurodegeneration induced by trimethyltin (TMT) administration. Int J Mol Sci. 2013 Aug 15;14(8):16817–16835. PubMed PMID: 23955266; PubMed Central PMCID: PMC3759937.
- Ceglia I, Reitz C, Gresack J, et al. APP intracellular domain–WAVE1 pathway reduces amyloid-β production. Nat Med. 2015;21(9):1054–1059.
- Bodero M, Bovee TFH, Wang S, et al. Screening for the presence of lipophilic marine biotoxins in shellfish samples using the neuro-2a bioassay. Food Addit Contam Part A Chem Anal Control Expo Risk Assess. 2018 Feb;35(2):351–365. PubMed PMID: 28884655.
- Chowanadisai W, Graham DM, Keen CL, et al. Neurulation and neurite extension require the zinc transporter ZIP12 (slc39a12). Proc Natl Acad Sci U S A. 2013;110(24):9903–9908.
- Wei PF, Zhang L, Nethi SK, et al. Accelerating the clearance of mutant huntingtin protein aggregates through autophagy induction by europium hydroxide nanorods. Biomaterials. 2014 Jan;35(3):899–907. PubMed PMID: 24169003.
- Areti A, Komirishetty P, Akuthota M, et al. Melatonin prevents mitochondrial dysfunction and promotes neuroprotection by inducing autophagy during oxaliplatin-evoked peripheral neuropathy. J Pineal Res. 2017 Apr;62(3):e12393. PubMed PMID: 28118492.
- Song Y, Du Y, Zou W, et al. Involvement of impaired autophagy and mitophagy in Neuro-2a cell damage under hypoxic and/or high-glucose conditions. Sci Rep. 2018;8(1). DOI:10.1038/s41598-018-20162-1
- Ktistakis NT, Tooze SA. Digesting the expanding mechanisms of autophagy. Trends Cell Biol. 2016 Aug;26(8):624–635. PubMed PMID: 27050762.
- Lawrence RE, Zoncu R. The lysosome as a cellular centre for signalling, metabolism and quality control. Nat Cell Biol. 2019 Feb;21(2):133–142. PubMed PMID: 30602725.
- Fraldi A, Klein AD, Medina DL, et al. Brain disorders due to lysosomal dysfunction. Annu Rev Neurosci. 2016 Jul 8;39(1):277–295. PubMed PMID: 27090953.
- Reuhl KR, Gilbert SG, Mackenzie BA, et al. Acute trimethyltin intoxication in the monkey (Macaca fascicularis). Toxicol Appl Pharmacol. 1985 Jul;79(3):436–452. PubMed PMID: 3929430.
- Little AR, Miller DB, Li S, et al. Trimethyltin-induced neurotoxicity: gene expression pathway analysis, q-RT-PCR and immunoblotting reveal early effects associated with hippocampal damage and gliosis. Neurotoxicol Teratol. 2012 Jan-Feb;34(1):72–82. PubMed PMID: 22108043.
- Saftig P, Klumperman J. Lysosome biogenesis and lysosomal membrane proteins: trafficking meets function. Nat Rev Mol Cell Biol. 2009 Sep;10(9):623–635. PubMed PMID: 19672277.
- Mindell JA. Lysosomal acidification mechanisms. Annu Rev Physiol. 2012;74(1):69–86. PubMed PMID: 22335796.
- Karle KN, Mockel D, Reid E, et al. Axonal transport deficit in a KIF5A(-/-) mouse model. Neurogenetics. 2012 May;13(2):169–179. PubMed PMID: 22466687; PubMed Central PMCID: PMCPMC3332386.
- Blakeslee WW, Lin YH, Stratton MS, et al. Class I HDACs control a JIP1-dependent pathway for kinesin-microtubule binding in cardiomyocytes. J Mol Cell Cardiol. 2017 Nov;112:74–82. PubMed PMID: 28886967; PubMed Central PMCID: PMCPMC5845792.
- Millecamps S, Julien JP. Axonal transport deficits and neurodegenerative diseases. Nat Rev Neurosci. 2013 Mar;14(3):161–176. PubMed PMID: 23361386.
- Maday S, Twelvetrees AE, Moughamian AJ, et al. Axonal transport: cargo-specific mechanisms of motility and regulation. Neuron. 2014 Oct 22;84(2):292–309. PubMed PMID: 25374356; PubMed Central PMCID: PMCPMC4269290.
- Ferguson SM. Axonal transport and maturation of lysosomes. Curr Opin Neurobiol. 2018 Aug;51:45–51. PubMed PMID: 29529416; PubMed Central PMCID: PMCPMC6066426.
- Hou J, Xue J, Lee M, et al. Ginsenoside Rd as a potential neuroprotective agent prevents trimethyltin injury. Biomed Rep. 2017 Apr;6(4):435–440. PubMed PMID: 28413642; PubMed Central PMCID: PMCPMC5374896.
- Geloso MC, Corvino V, Michetti F. Trimethyltin-induced hippocampal degeneration as a tool to investigate neurodegenerative processes. Neurochem Int. 2011 Jun;58(7):729–738. PubMed PMID: 21414367.
- Kim J, Son Y, Kim J, et al. Developmental and degenerative modulation of GABAergic transmission in the mouse hippocampus. Int J Dev Neurosci. 2015 Dec;47(Pt B):320–332. PubMed PMID: 26394279.
- Fiedorowicz A, Figiel I, Kaminska B, et al. Dentate granule neuron apoptosis and glia activation in murine hippocampus induced by trimethyltin exposure. Brain Res. 2001 Sep 7;912(2):116–127. PubMed PMID: 11532427.
- Yang M, Kim J, Kim T, et al. Possible involvement of galectin-3 in microglial activation in the hippocampus with trimethyltin treatment. Neurochem Int. 2012;61(7):955–962.
- McMillan DE, Wenger GR. Neurobehavioral toxicology of trialkyltins. Pharmacol Rev. 1985 Dec;37(4):365–379. PubMed PMID: 2872688.
- Dixit AB, Banerjee J, Srivastava A, et al. RNA-seq analysis of hippocampal tissues reveals novel candidate genes for drug refractory epilepsy in patients with MTLE-HS. Genomics. 2016 May;107(5):178–188. PubMed PMID: 27094248.
- Nakajima K, Yin X, Takei Y, et al. Molecular motor KIF5A is essential for GABA(A) receptor transport, and KIF5A deletion causes epilepsy. Neuron. 2012 Dec 6;76(5):945–961. PubMed PMID: 23217743.
- Rydzanicz M, Jagla M, Kosinska J, et al. KIF5A de novo mutation associated with myoclonic seizures and neonatal onset progressive leukoencephalopathy. Clin Genet. 2017 May;91(5):769–773. PubMed PMID: 27414745.
- Chen RF, Zhang T, Sun YY, et al. Oxygen-glucose deprivation regulates BACE1 expression through induction of autophagy in Neuro-2a/APP695 cells. Neural Regen Res. 2015 Sep;10(9):1433–1440. PubMed PMID: 26604904; PubMed Central PMCID: PMCPMC4625509.
- Pi H, Li M, Tian L, et al. Enhancing lysosomal biogenesis and autophagic flux by activating the transcription factor EB protects against cadmium-induced neurotoxicity. Sci Rep. 2017 Feb 27;7(1):43466. PubMed PMID: 28240313; PubMed Central PMCID: PMCPMC5327481.
- Liang Y, Huang M, Jiang X, et al. The neuroprotective effects of berberine against amyloid beta-protein-induced apoptosis in primary cultured hippocampal neurons via mitochondria-related caspase pathway. Neurosci Lett. 2017 Aug;10(655):46–53. PubMed PMID: 28668383.
- Qu M, Zhou Z, Chen C, et al. Lycopene protects against trimethyltin-induced neurotoxicity in primary cultured rat hippocampal neurons by inhibiting the mitochondrial apoptotic pathway. Neurochem Int. 2011 Dec;59(8):1095–1103. PubMed PMID: 22032970.
- Kuramoto N, Seko K, Sugiyama C, et al. Trimethyltin initially activates the caspase 8/caspase 3 pathway for damaging the primary cultured cortical neurons derived from embryonic mice. J Neurosci Res. 2011 Apr;89(4):552–561. PubMed PMID: 21290413.
- Dayton RD, Grames MS, Klein RL. More expansive gene transfer to the rat CNS: AAV PHP.EB vector dose-response and comparison to AAV PHP.B. Gene Ther. PubMed PMID: 30013186. Aug2018;25(5):392–400.
- Shin EJ, Nam Y, Tu TH, et al. Protein kinase Cdelta mediates trimethyltin-induced neurotoxicity in mice in vivo via inhibition of glutathione defense mechanism. Arch Toxicol Suppl. 2016 Apr;90(4):937–953. PubMed PMID: 25895139.
- Xu S, Pi H, Chen Y, et al. Cadmium induced Drp1-dependent mitochondrial fragmentation by disturbing calcium homeostasis in its hepatotoxicity. Cell Death Dis. 2013 Mar 14;4(3):e540. PubMed PMID: 23492771; PubMed Central PMCID: PMCPMC3615741.
- Wisniewski JR, Zougman A, Nagaraj N, et al. Universal sample preparation method for proteome analysis. Nat Methods. 2009 May;6(5):359–362. PubMed PMID: 19377485.
- Fan T, Pi H, Li M, et al. Inhibiting MT2-TFE3-dependent autophagy enhances melatonin-induced apoptosis in tongue squamous cell carcinoma. J Pineal Res. 2018 Mar;64(2):e12457. PubMed PMID: 29149494.
- Pi H, Xu S, Zhang L, et al. Dynamin 1-like-dependent mitochondrial fission initiates overactive mitophagy in the hepatotoxicity of cadmium. Autophagy. 2013 Nov 1;9(11):1780–1800. PubMed PMID: 24121705.
- Yang M, Pi H, Li M, et al. From the cover: autophagy induction contributes to cadmium toxicity in mesenchymal stem cells via AMPK/FOXO3a/BECN1 signaling. Toxicol Sci. 2016 Nov;154(1):101–114. PubMed PMID: 27492225.
- Wenzel ED, Speidell A, Flowers SA, et al. Histone deacetylase 6 inhibition rescues axonal transport impairments and prevents the neurotoxicity of HIV-1 envelope protein gp120. Cell Death Dis. 2019 Sep 12;10(9):674. PubMed PMID: 31515470; PubMed Central PMCID: PMCPMC6742654.
- Lorenzo DN, Badea A, Zhou R, et al. betaII-spectrin promotes mouse brain connectivity through stabilizing axonal plasma membranes and enabling axonal organelle transport. Proc Natl Acad Sci U S A. 2019 Jul 30;116(31):15686–15695. PubMed PMID: 31209033; PubMed Central PMCID: PMCPMC6681763.
- Farfel-Becker T, Roney JC, Cheng XT, et al. Neuronal soma-derived degradative lysosomes are continuously delivered to distal axons to maintain local degradation capacity. Cell Rep. 2019 Jul 2;28(1):51–64 e4. PubMed PMID: 31269450; PubMed Central PMCID: PMCPMC6696943.
- Liang C, Shao Q, Zhang W, et al. Smcr8 deficiency disrupts axonal transport-dependent lysosomal function and promotes axonal swellings and gain of toxicity in C9ALS/FTD mouse models. Hum Mol Genet. 2019 Oct 18;28(23):3940–3953. PubMed PMID: 31625563.
- Zuo G, Zhang T, Huang L, et al. Activation of TGR5 with INT-777 attenuates oxidative stress and neuronal apoptosis via cAMP/PKCepsilon/ALDH2 pathway after subarachnoid hemorrhage in rats. Free Radic Biol Med. 2019 Sep;4(143):441–453. PubMed PMID: 31493504.
- Cao Y, Li Q, Liu L, et al. Modafinil protects hippocampal neurons by suppressing excessive autophagy and apoptosis in mice with sleep deprivation. Br J Pharmacol. 2019 May;176(9):1282–1297. PubMed PMID: 30767208; PubMed Central PMCID: PMCPMC6468261.
- Vitner EB, Dekel H, Zigdon H, et al. Altered expression and distribution of cathepsins in neuronopathic forms of Gaucher disease and in other sphingolipidoses. Hum Mol Genet. 2010 Sep 15;19(18):3583–3590. PubMed PMID: 20616152.
- Toomey CB, Cauvi DM, Hamel JC, et al. Cathepsin B regulates the appearance and severity of mercury-induced inflammation and autoimmunity. Toxicol Sci. 2014 Dec;142(2):339–349. PubMed PMID: 25237059; PubMed Central PMCID: PMCPMC4250846.
- Sims-Robinson C, Bakeman A, Rosko A, et al. The role of oxidized cholesterol in diabetes-induced lysosomal dysfunction in the brain. Mol Neurobiol. 2016 May;53(4):2287–2296. PubMed PMID: 25976368; PubMed Central PMCID: PMCPMC4644712.
- Pi H, Li M, Zou L, et al. AKT inhibition-mediated dephosphorylation of TFE3 promotes overactive autophagy independent of MTORC1 in cadmium-exposed bone mesenchymal stem cells. Autophagy. 2019 Apr;15(4):565–582. PubMed PMID: 30324847.
- Xu S, Zhong M, Zhang L, et al. Overexpression of Tfam protects mitochondria against beta-amyloid-induced oxidative damage in SH-SY5Y cells. Febs J. 2009 Jul;276(14):3800–3809. PubMed PMID: 19496804.