
ABSTRACT
Autophagy is a highly conserved catabolic process and a major cellular pathway for the degradation of long-lived proteins and cytoplasmic organelles. An increasing body of evidence has unveiled autophagy as an indispensable biological function that helps to maintain normal tissue homeostasis and metabolic fitness that can also lead to severe consequences for the normal cellular functioning when altered. Recent accumulating data point to autophagy as a key player in a wide variety of physiological and pathophysiological conditions in the human endometrium, one of the most proficient self-regenerating tissues in the human body and an instrumental player in placental species reproductive function. The current review highlights the most recent findings regarding the process of autophagy in the normal and cancerous endometrial tissue. Current research efforts aiming to therapeutically exploit autophagy and the methodological approaches used are discussed.
Abbreviations: 3-MA: 3-methyladenine; ACACA (acetyl-CoA carboxylase alpha); AICAR: 5-aminoimidazole-4-carboximide riboside; AKT: AKT serine/threonine kinase; AMPK: AMP-activated protein kinase; ATG: autophagy related; ATG12: autophagy related 12; ATG16L1: autophagy related 16 like 1; ATG3: autophagy related 3; ATG4C: autophagy related 4C cysteine peptidase; ATG5: autophagy related 5; ATG7: autophagy related 7; ATG9: autophagy related 9; Baf A1: bafilomycin A1; BAX: BCL2 associated X, apoptosis regulator; BCL2: BCL2 apoptosis regulator; BECN1: beclin 1; CACNA1D: calcium voltage-gated channel subunit alpha1 D; CASP3: caspase 3; CASP7: caspase 7; CASP8: caspase 8; CASP9: caspase 9; CD44: CD44 molecule (Indian blood group); CDH1: cadherin 1; CDKN1A: cyclin dependent kinase inhibitor 1A; CDKN2A: cyclin dependent kinase inhibitor 2A; CMA: chaperone-mediated autophagy; CQ: chloroquine; CTNNB1: catenin beta 1; DDIT3: DNA damage inducible transcript 3; EC: endometrial cancer; EGFR: epidermal growth factor receptor; EH: endometrial hyperplasia; EIF4E: eukaryotic translation initiation factor 4E; EPHB2/ERK: EPH receptor B2; ER: endoplasmic reticulum; ERBB2: er-b2 receptor tyrosine kinase 2; ERVW-1: endogenous retrovirus group W member 1, envelope; ESR1: estrogen receptor 1; FSH: follicle-stimulating hormone; GCG/GLP1: glucagon; GFP: green fluorescent protein; GIP: gastric inhibitory polypeptide; GLP1R: glucagon-like peptide-1 receptor; GLS: glutaminase; H2AX: H2A.X variant histone; HIF1A: hypoxia inducible factor 1 alpha; HMGB1: high mobility group box 1; HOTAIR: HOX transcript antisense RNA; HSPA5: heat shock protein family A (HSP70) member 5; HSPA8: heat shock protein family A (HSP70) member 8; IGF1: insulin like growth factor 1; IL27: interleukin 27; INS: insulin; ISL: isoliquiritigenin; KRAS: KRAS proto-oncogene, GTPase; LAMP2: lysosomal-associated membrane protein 2; lncRNA: long-non-coding RNA; MAP1LC3A/LC3A: microtubule associated protein 1 light chain 3 alpha; MAP1LC3B/LC3B: microtubule associated protein 1 light chain 3 beta; MAPK8: mitogen-activated protein kinase 8; MAPK9: mitogen-activated protein kinase 9; MPA: medroxyprogesterone acetate; MTOR: mechanistic target of rapamycin kinase; MTORC1: mechanistic target of rapamycin kinase complex 1; MTORC2: mechanistic target of rapamycin kinase complex 2; MYCBP: MYC-binding protein; NFE2L2: nuclear factor, erythroid 2 like 2; NFKB: nuclear factor kappa B; NFKBIA: NFKB inhibitor alpha; NK: natural killer; NR5A1: nuclear receptor subfamily 5 group A member 1; PARP1: poly(ADP-ribose) polymerase 1; PAX2: paired box 2; PDK1: pyruvate dehydrogenase kinase 1; PDX: patient-derived xenograft; PIK3C3/Vps34: phosphatidylinositol 3-kinase catalytic subunit type 3; PIK3CA: phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha; PIK3R1: phosphoinositide-3-kinase regulatory subunit 1; PIKFYVE: phosphoinositide kinase, FYVE-type zinc finger containing; PPD: protopanaxadiol; PRKCD: protein kinase C delta; PROM1/CD133: prominin 1; PtdIns3K: class III phosphatidylinositol 3-kinase; PtdIns3P: phosphatidylinositol-3-phosphate; PTEN: phosphatase and tensin homolog; RB1CC1/FIP200: RB1 inducible coiled-coil 1; RFP: red fluorescent protein; RPS6KB1/S6K1: ribosomal protein S6 kinase B1; RSV: resveratrol; SGK1: serum/glucocorticoid regulated kinase 1; SGK3: serum/glucocorticoid regulated kinase family member 3; SIRT: sirtuin; SLS: stone-like structures; SMAD2: SMAD family member 2; SMAD3: SMAD family member 3; SQSTM1: sequestosome 1; TALEN: transcription activator-like effector nuclease; TGFBR2: transforming growth factor beta receptor 2; TP53: tumor protein p53; TRIB3: tribbles pseudokinase 3; ULK1: unc-51 like autophagy activating kinase 1; ULK4: unc-51 like kinase 4; VEGFA: vascular endothelial growth factor A; WIPI2: WD repeat domain, phosphoinositide interacting 2; XBP1: X-box binding protein 1; ZFYVE1: zinc finger FYVE domain containing 1.
Introduction
Autophagy (a term derived from Greek for “self-eating”) is a tightly regulated and highly conserved lysosomal degradation pathway. The autophagy process is induced under various conditions of cellular stress, resulting in the self-degradation of cellular components. Autophagy is broadly considered as a cell survival pathway. The process is required to preserve cell fitness through both nonselective and selective sequestration of macromolecules (proteins, lipids, glycogen, and nucleotides) and cellular organelles through a mechanism that involves the recognition of autophagy selective substrates by specific receptors [Citation1] to sustain metabolism and homeostasis [Citation2].
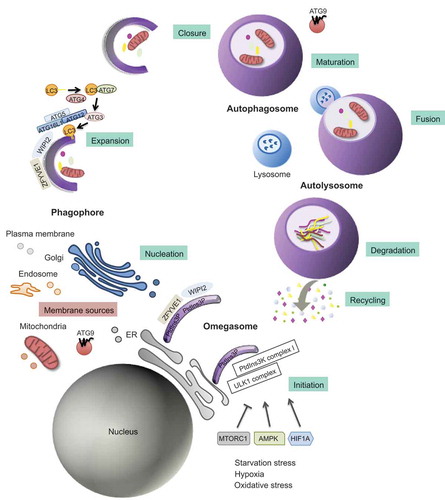
Three major autophagy routes with distinct molecular features have been described and extensively reviewed by other authors [Citation3–5]. These autophagy pathways are commonly referred to as chaperone-mediated autophagy (CMA), microautophagy, and macroautophagy [Citation6] (). Briefly, CMA is characterized by the direct delivery of single proteins into the lysosomal lumen through a pathway involving specific individual protein recognition by HSPA8 (heat shock protein family A [HSP70] member 8) cytosolic chaperone and subsequent lysosomal membrane translocation by interacting with LAMP2 (lysosomal-associated membrane protein 2) [Citation7]. Interestingly, all proteins internalized by the lysosomes through the CMA are characterized by a pentapeptide motif contained within their amino acid sequence (KFERQ). This pentapeptide is necessary for protein recognition by HSPA8 and lysosome targeting [Citation8]. Meanwhile, microautophagy is a process in which the lysosome directly engulfs the cytoplasmic material through the invagination of the lysosomal membrane [Citation9]. Among the different autophagy pathways, the most well-known and described route in the endometrial physiology is macroautophagy/autophagy. This form of autophagy is controlled by the highly conserved autophagy-related (ATG) genes [Citation10]. It is characterized by the sequestration of cytoplasmic portions within double-membraned vesicles known as autophagosomes. Subsequent fusion with the lysosomes generates de novo structures referred to as autolysosomes. The final degradation step is triggered by lysosomal hydrolases [Citation11]. These enzymes generate amino acids and macromolecules that are transported across the lysosomal membrane to the cytosol, where they are reutilized for anabolic processes [Citation12]. This multi-stage process can be divided into several steps: initiation, expansion, closure, and fusion with the endolysosomal system [Citation13] ().
Figure 1. Schematic representation of the autophagy process. Autophagy is a multi-stage process that comprises several steps: initiation, expansion, closure, and fusion with the endolysosomal system. The process initiates with the progressive sequestration of a portion of the cytoplasm that is enclosed within a double-membrane (a phagophore) to finally form an autophagosome. The initiation involves the transmission of the autophagy signal, which is triggered by multiple conditions, such as nutrient starvation, and canalized through specific cell sensors such as AMP-activated protein kinase (AMPK), HIF1A (hypoxia-inducible factor alpha) and mechanistic target of rapamycin kinase complex 1 (MTORC1) to the membrane of origin at which the nucleation takes place. As a consequence, there is a recruitment of the key initiation complexes. The ULK1 (unc-51 like autophagy activating kinase 1) complex plays a central role in the initiation stage, and it is the target of these signaling pathways and, when activated, triggers the nucleation of the phagophore by phosphorylating components of the class III phosphatidylinositol 3-kinase (PtdIns3k) complex I. Then, WIPI2 (WD repeat domain, phosphoinositide interacting 2), and ZFYVE1 (zinc finger FYVE domain containing 1) are recruited to the omegasome. WIPI2 binds and is activated by phosphatidylinositol 3-phosphate (PtdIns3P). Once activated, WIPI2 recruits at phagophore assembly sites the ATG12–ATG5-ATG16L1 complex that enhances the ATG3-mediated conjugation of LC3 (microtubule-associated protein light chain 3) to phosphatidylethanolamine. LC3-I is converted into its lipidated form, LC3-II. Membrane material for the nucleation of the phagophore and the elongation of the autophagic membrane arises from the ER, mitochondria, recycling endosomes, Golgi apparatus, and plasma membrane from which ATG9-containing vesicles provide portions of lipid bilayers. Finally, the expansion continues until the complete formation and closure of the autophagosome. The outer membrane of the autophagosome subsequently fuses with the lysosome, giving rise to the autolysosome. After lysosomal digestion, the sequestered cytoplasmic material is degraded into amino acids and macromolecules, transported across the lysosomal membrane to the cytosol, and finally recycled for anabolic processes
Box 1. Definitions
Functionally, autophagy is gaining momentum as a key regulator of multiple cellular functions due to its capacity to prevent the accumulation of cytotoxic products. Normal cellular metabolism, for instance, may generate damaged proteins or organelles and redox-active protein aggregates that require an intercellular quality control system, such as autophagy [Citation14]. In addition, autophagy responds to disturbances in intracellular or extracellular homeostasis, thus uncovering autophagy as a highly dynamic process, as evidenced by its role during mammalian development and differentiation [Citation15]. It is now apparent that autophagy deficiency or deregulation is related to various human diseases, such as cancer, as well as infectious, autoimmune, cardiovascular, rheumatic, pulmonary, metabolic diseases, obesity, aging, and neurodegenerative disorders (Alzheimer, Parkinson, and Huntington), and is thus a potential target for therapeutic mediation [Citation5].
In this article, we will review the current knowledge about the role of autophagy in the endometrium, which is an expanding area of research due to its key functions in reproductive biology and regeneration. Accordingly, only in recent years, multiple studies have pointed to autophagy as an important player in the normal endometrium physiological processes, as well as in pathological contexts, such as hyperplasia or endometrial cancer. These processes involve a constant adaptation to the ever-changing environment, cell growth, and apoptosis, intimately linked to autophagy. The current article reviews the most recent knowledge of the biological and pathological role of autophagy in the endometrium.
Autophagy in the normal physiology of the endometrium
The endometrium, or the innermost lining of the uterine cavity, is one of the most dynamic tissues in the human body. The other layers that compose the uterine wall are the myometrium or the middle layer, a muscular layer composed mainly by uterine smooth muscle, and the outermost serosal layer or perimetrium that covers the uterus.
Morphologically, the human endometrium is divided into two layers: the functional layer and the basal layer. The functional layer represents two-thirds of the endometrial thickness and comprises of different cellular components, including the glandular epithelium, endometrial stromal cells, fibroblasts, immunocompetent cells, and the vascular compartments [Citation16,Citation17]. Correct assembly of all these components concocts a dynamic tissue, which is under the control of estrogen and progesterone, that undergoes a complex series of cyclical changes during reproductive life in preparation for embryonic implantation and subsequent shedding and regeneration in the non-conceptive cycle [Citation18].
Menstrual cycle
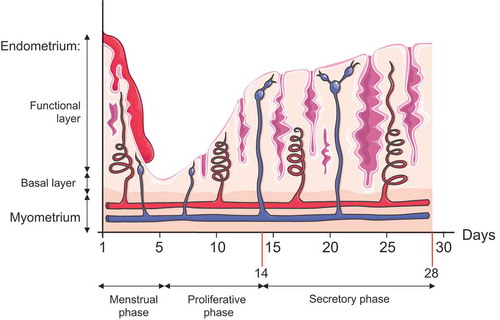
Distinct histologic phases are recognized during the menstrual cycle, which are: (i) menstrual phase, (ii) proliferative phase, and (iii) secretory phase (early secretory, mid-secretory, and late-secretory phases) (). The first day of menstrual bleeding corresponds to the beginning of the endometrial cycle and results from the reduction in estrogen and progesterone concentrations. This hormone reduction leads to a constriction of spiral arteries and ischemic necrosis of the functional endometrium layer. During the late-secretory phase and the menstrual phase, apoptosis seems to facilitate the elimination of the senescent endometrial cells from the functional layer of the endometrium [Citation19]. This phase is followed by the proliferative phase, characterized by the progressive increase in the synthesis and secretion of estradiol and concomitant thickening of the endometrium. Then, during the secretory phase, the ovaries produce progesterone [Citation20], which plays an important role in preparing the endometrium for the blastocyst implantation by boosting blood flow, uterine secretions, and by reducing the contractility of the smooth muscle layer. In the absence of implantation, withdrawal of estrogen and progesterone leads to the vasoconstriction of spiral arteries, resulting in the involution of the endometrium and restarting of the menstrual cycle. In this context, Choi et al. demonstrated that endometrial cell autophagy is controlled by ovarian steroids and closely correlates with apoptosis during the secretory and menstruating phases. Specifically, the authors observed increased levels of lipidated MAP1LC3A/LC3A (microtubule associated protein 1 light chain 3 alpha) or LC3A-II, which peaks in the late-secretory phase and correlates with cleaved CASP3 (caspase 3). Autophagy and apoptosis present a close relationship during the menstrual cycle [Citation21]. Importantly, the authors also demonstrated that ovarian hormones regulate autophagy in endometrial cancer cells in vitro. LC3A-II levels increase in Ishikawa cells deprived of estrogen and/or progesterone. The autophagy inhibitor bafilomycin A1 (Baf A1), promotes autophagosome accumulation and increases the BAX:BCL2 (BCL2 associated X, apoptosis regulator:BCL2 apoptosis regulator) ratio and cleaved CASP3 thereby, triggering an apoptotic response.
Figure 2. Illustration of endometrial layers and tissue components dynamic changes during the course of the different phases of the menstrual cycle. The endometrium, composed of a single layer of columnar epithelial cells plus stroma, can be subdivided into two layers: the functional, which faces the uterine cavity, and the basal layer in direct contact with the myometrium. Both layers undergo structural changes during the menstrual cycle. Menstruation constitutes the first phase of the menstrual cycle, and it is characterized by the complete desquamation of the functional layer of the endometrium and bleeding (menses). This stage precedes the proliferative phase, characterized by the increase in the synthesis and secretion of estrogens as the ovarian follicles mature (ovarian follicular phase). The estrogens will promote the regeneration of the functional layer from the basal layer of the endometrium. By day 14, a peak in LH secretion triggers a decrease in estrogen production, ovulation, and the formation of the corpus luteum (luteal ovarian phase), which represents the primary source of progesterone. Progesterone will prepare the endometrium for future implantation by fostering global endometrial thickening characterized by increased spiral artery length and coiling, uterine secretions, and reduced smooth muscle cell contractility. In the absence of pregnancy, the demise of the corpus luteum will cause a dramatic drop in the progesterone and estrogen levels, and the initiation of the menstruation starts again
Implantation
Implantation is a coordinated process involving a functional blastocyst, a receptive endometrium, a synchronized transformation, and an adequate cross-communication between embryonic and maternal tissues. The process, ultimately, results in the blastocyst adherence to the uterine wall and nourishment [Citation22] ().
Box 2. Definitions
During implantation, the endometrium must suffer several adaptations to ensure the development of a proper feto-maternal interface. Changes in the uterine mucosa are known as decidual reaction, decidualization, or transformation into a decidua. Decidualization is characterized by the differentiation and conversion of the endometrial stromal cells into epithelioid-like cells to accommodate pregnancy [Citation23] (). Both resultant specialized secretory decidual cells that supply nutrients and an immune-favorable microenvironment are necessary for successful implantation and placentation [Citation24]. This process occurs in the area where the blastocyst and the endometrial decidua contact and requires a postovulatory increase of progesterone and local cAMP levels [Citation24]. It involves swelling of all endometrial stromal cells, due to the accumulation of lipid granules, glycogen, and other nutrients, to enable the survival of the embryo until the placenta is completely developed [Citation23]. The process of epithelioid differentiation that occurs during decidualization also involves an increase in the complexity and number of the nucleoli, accumulation of lipid droplets and glycogen into the cytoplasm, and an increase and dilatation of rough ER (endoplasmic reticulum) and Golgi apparatus [Citation25,Citation26].
Interestingly, recent pieces of evidence suggest that autophagy favors decidualization as indicated by the remarkable presence of MAP1LC3/LC3 (microtubule-associated protein light chain 3/LC3-I), converted LC3-II expression, and autophagosomes in normal, abnormal villous and decidual specimens from early pregnancies [Citation27]. An additional study recently demonstrated that autophagic flux is increased during decidualization using an immortalized cell line of human endometrial stromal cells. Importantly, knockdowns of ATG7 and ATG5 impair decidualization, confirming the role of these ATG proteins in the human endometrial decidualization [Citation28]. In support of this, Rhee et al. used an obese mouse model to demonstrate that impaired autophagy is linked to a decrease in decidualization and obesity-related infertility [Citation29]. Obesity has been shown to exert negative effects on reproduction and correlated with decreased pregnancy rates, infertility [Citation30], increased early pregnancy loss [Citation31], and poor oocyte quality [Citation32]. Rhee et al. demonstrated that diet-induced obesity reduces endometrial stromal cell decidualization, implantation, and early fetal growth [Citation29]. Interestingly, the authors observed that autophagy is induced during decidualization, and characterized by increased levels of ACACA (acetyl-CoA carboxylase alpha), p-ACACA and p-ULK1 (unc-51 like autophagy activating kinase 1). These autophagy markers are significantly decreased in decidualizing cells from obese females. Not surprisingly, the authors reported a higher autophagic flux in decidualized cells since the expression of LC3B-II increases with decidualization in human endometrial stromal cells exposed to Baf A1. This effect is abolished after palmitic acid exposure. The results point at an intriguing link between autophagy and obesity in the context of reproductive fitness. Autophagy inducers may provide a potential therapeutic for the treatment of obesity-associated subfertility.
Noteworthy, a role for autophagy has also been demonstrated in the context of oocytogenesis, embryogenesis, implantation, and placentation. Disruption of autophagy has been linked to spontaneous abortion, poor placentation, pre-eclampsia, and intrauterine growth restriction [Citation33].
Atrophic endometrium
Menopause is characterized by an insufficient stimulation of the endometrium in response to a primary ovarian failure and a dramatic decrease in circulating estrogen (). The endometrium may become thinner as a result of low estrogen levels and is described as being atrophic. Autophagy has been described as a potential molecular mechanism responsible for estrogen withdrawal-induced endometrium atrophy [Citation34,Citation35]. Indeed, autophagy is activated in the uterine epithelial cells of both oophorectomized rats and peri-menopausal women and correlated with apoptosis [Citation34]. After oophorectomy, the uterine endometrium of rats becomes atrophic in a time-dependent manner. ER stress precedes this response by inactivating the AKT (AKT serine/threonine kinase)-MTOR (mechanistic target of rapamycin kinase) signaling pathway in vitro and in vivo through the inhibition of the arachidonic acid-prostaglandin E2 axis. These events result in increased autophagosomes and LC3-dependent autophagy in the atrophic endometrium. Similarly, the exposure of primary epithelial cells to the estrogen inhibitor, letrozole, leads to elevated autophagic flux and apoptotic response characterized by increased BAX, BCL2, and active CASP3 protein levels. Importantly, administration of exogenous 17β-estradiol, prostaglandin E2, or salubrinal, and silencing of ATG5 and BECN1 (beclin 1) counterbalance these effects. Tissue-level analysis demonstrated that, while premenopausal uterine epithelial cells present low autophagy level and death, apoptosis in postmenopausal uterine epithelial cells is concomitant with autophagy. Altogether, these findings point to autophagy and estrogen withdrawal as regulators of the endometrial atrophy and suggest that autophagy may induce apoptotic cell death.
The transition to menopause is characterized by an increase in serum FSH (follicle-stimulating hormone), and a decrease in estradiol and inhibin levels [Citation36]. Recent pieces of evidence demonstrate that FSH participates in age-related endometrial atrophy, partly through regulation of the autophagy pathway [Citation35]. Surgical oophorectomy in mice results in endometrium alterations, such as smaller glandular tube size, vacuolization in mitochondria, pyknotic nuclei, and a hollowed rough ER. In line with this, FSH inhibits cell proliferation and promotes apoptosis of primary endometrial cells. FSH increases CASP3, CASP8 (caspase 8), and CASP9 (caspase 9) apoptosis markers and the protein expression of the autophagy-related genes ATG3, ATG5, ATG7, ATG12 and LC3A/B in endometrial adenocytes. Additional mechanistic studies showed that the autophagy-related genes are transcriptionally regulated by the FSH-TGFBR2 (transforming growth factor beta receptor 2)-SMAD2 (SMAD family member 2)-SMAD3 signaling axis.
Autophagy in the pathologic endometrium
Hyperplasia
Endometrial hyperplasia (EH) is defined by the abnormal thickening of the endometrium. It is characterized by the irregular proliferation of endometrial glands and an increase in the gland-to-stroma ratio in comparison to the proliferative endometrium [Citation37]. EH comprises both benign and/or premalignant lesions since certain atypical forms are considered a precursor lesion of endometrial cancer (EC) [Citation38,Citation39]. Therefore, an accurate diagnosis that can discriminate between these two situations is essential for optimal patient management. Two different, yet overlapping, histomorphological systems are used to classify premalignant endometrial lesions: i) the World Health Organization (WHO) system of endometrial hyperplasia with or without atypia, which differentiates a high-risk from a low-risk of synchronous or subsequent adenocarcinoma based on the presence or absence of cytologic atypia and ii) the endometrial intraepithelial neoplasia system defined as a clonal proliferation of architecturally or cytologically altered endometrial glands [Citation40].
Chronic exposure of the endometrium to excessive or unopposed estrogen stimulation may result in EH. A well-known paradigm of treatment-induced EH is the chronic exposure to tamoxifen, a selective ESR1 (estrogen receptor 1) modulator. Tamoxifen is widely used in pre-menopausal women with ESR1-positive breast cancer, and it is the standard treatment in post-menopausal women with breast cancer. However, while presenting ESR1 antagonistic effects in the breast, tamoxifen exerts agonistic effects in other tissues, such as the uterus. Indeed, long-term tamoxifen treatment increases the incidence of EH and endometrial cancer [Citation41]. Additionally, tamoxifen has been shown to activate the transcription of several ESR1 target genes in endometrial cells, such as PAX2 (paired box 2), ERVW-1 (endogenous retrovirus group W member 1, envelope) [Citation42] and NR5A1 (nuclear receptor subfamily 5 group A member 1) [Citation43]. Tamoxifen promotes EH by activating PRKCD (protein kinase C delta) and by inducing NFE2L2 (nuclear factor, erythroid 2 like 2) phosphorylation at serine 40. Upon phosphorylation, NFE2L2 translocates into the nucleus and leads to increased transcription of the autophagy-related receptor SQSTM1 (sequestosome 1) [Citation41]. Interestingly, tamoxifen-mediated tumor growth is attenuated upon SQSTM1 knockdown in RL95-2 and AN3CA endometrial cancer cells but not in breast cancer cells. In vivo, a chemical inhibitor of NFE2L2 reverts tamoxifen-induced hyperplasia in endometrial cells but not in breast cancer cells.
Other complementary studies also linked autophagy to EH. For instance, Sivridis et al. studied the LC3A reactivity by immunohistochemistry in a series of endometrial cancer, hyperplasia, and normal endometrium [Citation44]. LC3A reactivity was characterized by three staining patterns: i) diffuse cytoplasmic, ii) cytoplasmic/juxta-nuclear, and iii) spheroidal structures enclosed within the cytoplasmic vacuoles, described as “stone-like” structures (SLS). Both cytoplasmic diffusion and juxta-nuclear patterns are associated with basal autophagy activity. The SLS pattern is observed in malignant epithelial cells, reflecting an increased or an abnormal autophagy level. Interestingly, the LC3A-positive SLS staining is also present in a small fraction of atypical hyperplasias. The authors suggested that the SLS counts could be a potential diagnostic tool to differentiate grade-1 endometrioid adenocarcinomas from atypical hyperplasias. The study suggested that autophagy is altered in endometrial hyperplasias and specifically increased in atypical hyperplasias.
Endometrial cancer
Endometrial cancer (EC) is the most common gynecologic malignancy in the Western world, with more than 320,000 estimated new cases worldwide in 2012 [Citation45,Citation46]. EC incidence has increased by 21% since 2008, probably related to the increasing life span and obesity rates [Citation47]. In 1983, Bokhman et al. described a dualistic model of endometrial cancer based on clinical, pathological, and molecular features [Citation48]. This model broadly categorizes EC to type I and type II tumors and has been used to classify EC up to present. Type I ECs account for ~75% of diagnosed ECs, typically representing low-grade estrogen-dependent tumors that mostly develop in perimenopausal women and are frequently associated with hyperplastic precursor lesions. On the other hand, type II ECs tend to be estrogen-independent tumors, with most cases occurring in postmenopausal women. They are aggressive tumors with a poorer clinical outcome and include mostly serous and clear cell histological subtypes [Citation49]. Contrary to type I, type II ECs are more frequently associated with endometrial atrophy. In addition to the clinical and pathological aspects, the differences between type I and type II tumors can be partially attributed to divergent alterations at a molecular level. While type I tumors present mutations in PTEN (phosphatase and tensin homolog), KRAS (KRAS proto-oncogene, GTPase), PIK3CA (phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha) and CTNNB1 (catenin beta 1), as well as microsatellite instability, type II tumors show alterations in TP53 (tumor protein p53), loss of heterozygosity on several chromosomes, as well as other representative molecular alterations (CDH1 [cadherin 1], ERBB2 [er-b2 receptor tyrosine kinase 2] and CDKN2A [cyclin dependent kinase inhibitor 2A]) [Citation50,Citation51].
Although the dualistic classification has been broadly used, it is not entirely robust since some endometrial cancers present shared characteristics of both types I and type II ECs. Recently, a new molecular classification of EC profiling has shed light on the different genomic subtypes and may facilitate the development of tailored treatments and improved patient management [Citation52].
Autophagy in endometrial cancer development
The aforementioned study by Sivridis et al. [Citation44] found that the number of SLS is significantly higher in EC specimens, compared to hyperplasias or healthy tissues. Contrary to the diffuse and the juxta-nuclear staining, which are related to a basal level of autophagy and low malignant potential, SLS-positive staining is associated with highly aggressive tumors, exaggerated autophagy levels, and a dramatic decrease in the 5-year survival. The authors stratified EC cases by favorable and unfavorable prognosis, revealing a first group largely represented by a low-grade endometrioid adenocarcinoma and the second one composed of serous, clear cell, and grade 2–3 endometroid ECs. Intriguingly, even in the high-grade tumors group, the presence of a high SLS count is associated with significantly worse prognosis, underscoring a potential role for autophagy in tumor aggressiveness.
More recently, another study examined the data from The Cancer Genome Atlas from 11 cancer types to analyze a set of 211 human autophagy-related genes establishing correlations with tumor-associated DNA alterations and RNA expression [Citation53]. Lebovitz et al. excellently reported EC as the tumor type presenting the highest number of significantly autophagy-associated mutated genes. Mutations include core autophagy genes (ATG4 C [autophagy related 4 C cysteine peptidase], ULK4 [unc-51 like kinase 4], and RB1CC1/FIP200 [RB1 inducible coiled-coil 1]), along with MTOR-activating mutations. Interestingly, practically all of the mutations are found in patients of the endometrioid subtype and include patients presenting double mutants for MTOR and PTEN, and patients presenting either a truncating mutation or a predicted damaging substitution in the autophagy inducer RB1CC1. These results imply that autophagy execution may be compromised in endometrioid tumors as a result of hyperactivation of MTOR, or inactivating mutations in core autophagy components such as RB1CC1. In contrast, non-endometrioid tumors exhibit an increase in mRNA levels of CDKN2A, an autophagy activator [Citation54], and a biomarker that is highly expressed in serous compared to endometrioid EC [Citation55].
Aberrant expression of other autophagy core components has also been reported in EC. For instance, increased BECN1 expression has been observed in endometrioid EC and associated with poor prognostic factors, such as high tumor grade, myometrial invasion, and low estimated 5-year survival [Citation56]. A positive correlation between BECN1 and HIF1A (hypoxia inducible factor 1 alpha) levels suggests a role for BECN1 in tumor survival in hypoxic microenvironments. In contrast to these findings, another study observed a decrease in BECN1 and PTEN from normal endometria throughout the neoplastic transformation, including hyperplasia and carcinoma transition [Citation57]. However, in this case, the expression of BECN1 has been correlated only with cell differentiation and histological type. No correlations with the myometrial infiltration or the pathologic stage of the tumor have been reported.
Another molecule linked to EC and autophagy is KIAA1324/EIG121, a transmembrane protein associated with intracellular membrane trafficking, endosomes, and autophagy activation [Citation58]. KIAA1324 is highly expressed in complex hyperplasia and low-grade endometrioid tumors but decreased in non-endometroid tumors [Citation59]. KIAA1324 overexpression in vitro increases autophagy levels, as observed by the accumulated autophagosomes, LC3 punctuated dot-like structures, acidic vesicles, and long-lived protein lysosomal degradation. In contrast, KIA1324 knockdown decreases cell survival during starvation or stress by using cytotoxic agents, suggesting its role in protecting cancer cell survival under hostile conditions through autophagy induction [Citation60].
In conclusion, a vast amount of evidence suggests that autophagy may contribute to EC development by enabling cell survival in unfavorable conditions, like starvation or hypoxia. In addition, although further studies are required, high CDKN2A mRNA levels and SLS counts in the more aggressive non-endometrioid subtype may indicate higher autophagy induction. In comparison, the endometrioid subtype may exhibit a lower level of autophagy competence due to genome DNA mutations.
Targeting autophagy as a therapeutic approach in endometrial cancer
EC generally presents a favorable prognosis due to its early detection, when primary surgical treatment is frequently curative, reaching 5-year survival rates over 70%. Despite being treated according to their assessed risk, ~15-20% of these early-diagnosed patients continue to relapse after surgery. Recurrences typically affect the vaginal or pelvic regions and may include metastasis to distant sites [Citation61,Citation62]. Consequently, patients diagnosed at the advanced-stages or patients presenting high-risk factors receive a more aggressive treatment after primary surgery. Treatments are based on adjuvant radiation therapy, chemotherapy, or the combination of both. However, these treatments tend to fail and are estimated to benefit only 10–15% of all patients [Citation63]. The prognosis of patients with recurred or advanced (metastatic) EC is consequently poor, with median survival rates of less than 1 year and a median progression-free survival rates of 4 months [Citation64]. These clinical features underscore the need to develop improved targeted therapies and novel strategies to overcome cancer resistance [Citation65]. Several efforts have been made to determine the potential therapeutic benefit of autophagy modulation as a novel treatment for EC. Over the last decade, the number of studies assessing the role of autophagy during EC response to therapy has grown exponentially. While numerous articles report a role for autophagy in response to multiple agents, these publications do not follow common, systematic, and standardized protocols to study autophagy. Major research findings involving the use of potential therapeutic drug compounds to treat EC (mechanism of action and autophagy modulation) are summarized in .
Table 1. Targeting autophagy in endometrial cancer
Currently, systemic therapy for EC mainly entails the use of platinum-based and taxane compounds. However, available options after the first-line treatment when cancer recurs are limited, thus evidencing the need for novel therapeutic drugs to overcome EC chemoresistance [Citation66]. Although the role of autophagy in cancer is complex and context-dependent, increasing pieces of evidence suggest that chemotherapeutic agents may activate an autophagy pro-survival response in cancer cells [Citation67,Citation68]. Inhibition of autophagy may re-sensitize chemoresistant cancer cells to conventional therapy. Importantly, autophagy inhibition increased cell sensitivity in response to a lower drug concentration and, consequently, may help reduce side effects. Conversely, chemotherapeutic agents have also been shown to trigger an autophagy response that leads to cell death, mainly in apoptosis-defective cells [Citation69]. This notion suggests a therapeutic strategy based on an over-stimulation or excessive autophagy. Further characterization of the autophagy-dependent adaptive mechanisms in cancer cells is crucial to overcome drug resistance and to improve drug efficacy.
Cisplatin and combination therapies
Chloroquine (CQ) is an autophagy inhibitor that increases the lysosomal pH. CQ treatment blocks the fusion of autophagosomes with lysosomes and subsequent lysosomal protein degradation, thereby promoting autophagosome accumulation [Citation70]. In Ishikawa, KLE, and AN3CA cell lines, CQ treatment increases the accumulation of LC3-II and SQSTM1 and suppresses cell proliferation via the induction of apoptosis [Citation71]. These CQ effects, then, decrease after ATG5 or ATG7 silencing. It was shown that cisplatin induces autophagy in Ishikawa cells, and either ATG5 or ATG7 knockdown significantly increases cell sensitivity to cisplatin. Furthermore, the combination of cisplatin and CQ in cisplatin-resistant cells enhances their sensitivity to cisplatin. These results indicate that the induction of autophagy protects cancer cells against cancer treatment.
Similarly, Sun M et al. reported that Ishikawa cisplatin-resistant cells present higher autophagy levels compared to cisplatin-sensitive parental cells [Citation72]. The authors performed long-non-coding RNA (lncRNA) profiling to identify differentially expressed lncRNAs involved in the regulation of cisplatin-induced autophagy. Among the differentially expressed lncRNAs, HOTAIR (HOX transcript antisense RNA) is significantly downregulated in resistant cells. Interestingly, the authors found that HOTAIR regulates the expression of BECN1. Downregulation of HOTAIR by siRNA increases autophagy and apoptosis in Ishikawa cisplatin-resistant cells. Remarkably, a dual-color DsRed-LC3-GFP reporter was used to study the increased autophagic flux, representing one of the few studies in EC using the state-of-the-art methodology. However, despite evidence of increased autophagic flux in resistant cells, no GFP+, RFP+, or double GFP+ RFP+ puncta per cell were quantified. The protective role of autophagy in this context is still unclear, as cell survival and autophagic flux were not evaluated upon autophagy inhibition using conventional agents (e.g., CQ) and upon decreased expression of HOTAIR, respectively.
Finally, another group found that cisplatin exposure in Ishikawa EC cells induces an autophagy response, with the increased number of autophagosomes and LC3 expression through inactivation of the PIK3CA-AKT-MTOR signaling pathway [Citation73]. This inhibition is partially reversed by co-treatment with IGF1 (insulin like growth factor 1), a PIK3CA activator.
Proteasome inhibitors
An additional class of compounds that affect autophagy levels are proteasome inhibitors. One example is bortezomib, an inhibitor of the 26S proteasome used in the treatment of multiple myeloma [Citation74]. Bortezomib also presents anti-tumoral effect in other malignancies, such as endometrial [Citation75], breast [Citation76], colon [Citation77], and prostate cancer [Citation78]. It has been described that bortezomib, by stabilizing NFKBIA (NFKB inhibitor alpha), blocks the activity of the NFKB (nuclear factor kappa B) signaling pathway, resulting in the downregulation of its anti-apoptotic target genes, increasing the sensitivity of cancer cells to chemotherapy [Citation79]. Treatment with CQ or bortezomib increases SQSTM1 and LC3-II in ovarian, endometrial, and hepatocellular cancer cells [Citation80]. However, bortezomib blocks the autophagic flux without inhibiting the fusion of autophagosomes with lysosomes, which may be due to suppressed cathepsin activities by EPHB2/ERK (EPH receptor B2) phosphorylation.
Histone deacetylase inhibitors
Histone acetylation modifications are responsible for the changes in the chromatin structure and the accessibility of genes for the transcriptional machinery and have a crucial role in epigenetic regulation of gene expression. Histone deacetylase inhibitors are considered as potential therapeutic agents in cancer treatment [Citation81], including EC [Citation82]. Suppression of sirtuin (SIRT) proteins and signaling by the novel SIRT inhibitor, MHY2256, results in cell cycle arrest in vitro and impaired tumor growth in a tumor xenograft mouse model of Ishikawa cells. MHY2256 induces early and late-stage apoptosis and autophagy, as shown by an increase in the levels of LC3-II, ATG5, and acidic vesicular organelles. MHY2256 exerts its antitumorigenic properties through TP53 acetylation.
Enhanced NK cytotoxic activity
Other studies have elegantly assessed the effects of cisplatin and autophagy modulation in the tumor microenvironment. For instance, a recent study suggested that rapamycin and cisplatin co-therapy may improve EC treatment by stimulating cytotoxicity of natural killer (NK) cells [Citation83]. NK cells are a type of innate cytotoxic lymphoid cells and are critical for controlling neoplastic development, tumor growth, metastatic spread, and determining tumor chemosensitivity [Citation84]. Zhou et al. reported a positive correlation between rapamycin treatment and increased IL27 (interleukin 27) expression, which impacts the degree of tumor differentiation. By implementing a co-culture system, the authors demonstrated that overexpression of IL27 or treatment with rapamycin in Ishikawa, RL95-2, and KLE cells enhances the cytotoxic activity of NK cells. This impairs EC tumor growth in a xenograft mouse model. Mechanistically, IL27 enhances rapamycin-induced autophagy through the upregulation of BECN1 and LC3B mRNA levels [Citation83]. Rapamycin combined with a classical cytotoxic agent (e.g., cisplatin) might be an improved therapy for EC, particularly in patients with poorly differentiated carcinomas or abnormally low expression levels of IL27.
Carboplatin, paclitaxel and combination therapies
Based on its response rates, the combination of carboplatin and paclitaxel is the standard regimen of EC treatment [Citation66,Citation85,Citation86]. A recent study pointed to ABTL0812, a novel class of chemically modified fatty acid-derived small molecule, as an effective compound acting on the PIK3CA-AKT-MTORC1 (mechanistic target of rapamycin kinase complex 1) axis [Citation87]. ABTL0812 induces autophagy-mediated cell death by inhibiting the AKT-MTOR axis through TRIB3 (tribbles pseudokinase 3) upregulation, an endogenous AKT inhibitor. The molecule also induces ER stress in vitro and in vivo. ABTL0812 reduces cell viability and increases cell death in Ishikawa, AN3CA, HEC-1A, ARK1, and ARK2 EC cell lines (CASP9 and CASP3). Concomitant with upregulated TRIB3 expression and reduced phosphorylation of AKT and RPS6KB1 (ribosomal protein S6 kinase B1), ABTL0812 increases the levels of LC3-II and the autophagic flux, quantified by using a chimeric mRFP-GFP tandem fluorescent-tagged LC3B construct (tfLC3). In addition, ABTL0812 impairs tumor progression in patient-derived xenograft (PDX) EC models (endometrioid and serous) and presents a similar efficacy to the standard first-line treatment (carboplatin-paclitaxel). The authors further demonstrated that ABTL0812 has a synergistic effect when combined with carboplatin-paclitaxel in the serous PDX model. Using an inducible pten knockout mice model, which carries Pten-floxed alleles (Ptenfl/fl), and the tamoxifen-induced CRE recombinase (Cre:Ers+/−) under the estrogen promoter, the authors observed that ABTL0812 treatment impairs pten knockout cells to progress from hyperplasia to cancer. Interestingly, ABTL0812 reduces proliferation and induces autophagy exclusively in pathogenic but not in healthy endometrial cells using 3D cultures. A phase 1b/2a clinical trial for the evaluation of ABTL0812 in patients with EC is ongoing (NCT03366480).
In another study, paclitaxel resistance in EC was tested using HEC-1A, JEC, Ishikawa, and AN3CA EC cell lines. HEC-1A and JEC cells, respectively, present the highest IC50 values and are considered paclitaxel-resistant cells [Citation88]. Paclitaxel induces autophagy in paclitaxel-insensitive HEC-1A and JEC cells. It increases the LC3-II:LC3-I ratio, LC3 punctate dots in the cytosol, BECN1 levels, and decreases SQSTM1 but not in the paclitaxel-sensitive Ishikawa and AN3CA cell lines. Treatment with both paclitaxel and CQ, or BECN1 shRNA, enhances cell death. This result suggested that paclitaxel-induced autophagy in EC-resistant cells plays a cytoprotective role. Mechanistically, paclitaxel stimulates the generation of reactive oxygen species, that, when inhibited, blocks paclitaxel-induced autophagy. Therefore, an autophagy inhibitor treatment in combination with paclitaxel may be an effective treatment in EC.
Ran et al. identified the MIR218 as significantly downregulated in Ishikawa and RL95-2 paclitaxel-resistant cells. MIR218 inversely correlates with the increased autophagy levels, indicated by accumulation of LC3 puncta, high LC3-II:LC3-I ratio, and BECN1 expression [Citation89]. Overexpression of MIR218 re-sensitizes resistant EC cells to paclitaxel by interacting with the 3ʹ-UTR of HMGB1 (high mobility group box 1). Accordingly, HMGB1 knockout by using transcription activator-like effector nuclease (TALEN) technology decreases the autophagy levels in paclitaxel-treated cells.
On the contrary, another study found that treatment with RAD001 (everolimus), an inhibitor of MTOR, induces autophagy-dependent cell death in Ishikawa and HEC-1A EC cells, and increases their sensitivity to low-dose paclitaxel [Citation90]. This result suggests that the combination of RAD001 with paclitaxel may reduce the cytotoxic effect of the chemotherapeutic. Treatment with RAD001 inhibits MTOR and RPS6KB1 phosphorylation, increasing the formation of autophagosomes and the expression of LC3-II in Ishikawa and HEC-1A cells. Moreover, both treatment with CQ or ATG5 shRNA knockdown inhibits RAD001-induced cell death. Altogether, these data indicate that RAD001 may potentially be used for treating EC patients that present hyperactivated AKT-MTOR signaling.
Cancer stem cells
Another major contributor to chemoresistance and recurrence is the presence of cancer stem cells within the tumor. Although the relationship between cancer stem cells and autophagy has been explored, a complete understanding of the context of EC is lacking. The first key step toward this line of research was recently undertaken by Ran X et al. [Citation91]. Endometrial PROM1/CD133 (prominin 1)-positive and CD44 (CD44 molecule [Indian blood group])-positive (PROM1+ CD44+) JEC cells present higher levels of autophagy, increased BECN1, ATG5, ATG7 mRNA, and LC3-II protein levels. Furthermore, the authors demonstrated that autophagy increases during serial passages of spheroid cultures in vitro and that autophagy inhibition by using CQ decreases stemness. Knockdown KIAA1324 reduces autophagy levels, as well as the stemness and the tumorigenic potential of PROM1+ CD44+ EC cells.
Metabolism
Metabolic reprogramming is a hallmark of cancer. Several changes in cellular bioenergetics of cancer cells allow them to adapt to adverse conditions. Cells reprogram metabolic pathways in order to survive and proliferate under nutrient-restrictive conditions. Metabolic reprogramming also alters cellular resistance to hypoxia, oxidative stress, and promotes immunological scape [Citation92].
Metformin is a biguanide hypoglycemic drug commonly used for the treatment of patients with type-2 diabetes. It works by decreasing the blood glucose levels and by increasing sensitivity to insulin of body tissues. Interestingly, metformin also reduces the proliferation of cancer cells and improves patient survival [Citation93]. It is also associated with a decreased incidence of cancer in diabetic patients [Citation94]. Metformin acts through direct activation of the AMP-activated protein kinase (AMPK), and inhibition of the MTOR pathway [Citation95,Citation96]. Metformin treatment in Ishikawa cells reduces proliferation through caspase-dependent apoptosis and cell-cycle arrest at G0/G1 and G2/M by activating CDKN1A (cyclin dependent kinase inhibitor 1A) via a TP53-independent pathway [Citation97]. Moreover, metformin induces autophagy activity, by increasing the ratio LC3-II:LC3-I, SQSTM1 degradation, and the formation of acidic vesicular organelles. Not surprisingly, inhibition of metformin-induced autophagy, by 3-methyladenine (3-MA) treatment or BECN1 siRNAs, reduces apoptosis in Ishikawa EC cells. These results point to metformin as a promising agent in the treatment of early EC.
The pharmacological modulation of key enzymatic activities, such as GCG/GLP1 (glucagon), has also been shown to impact autophagy in endometrial cells. Together with GIP (gastric inhibitory polypeptide), GCG represents an incretin that decreases blood glucose levels by enhancing the secretion of INS (insulin) [Citation98] and suppression of glucagon secretion and appetite. As opposed to GIP, GCG activity is preserved in patients with type-2 diabetes, and GCG analogs are a primary glycemic control agent for this pathology. The impact of liraglutide, a derivative of GCG that is used as a long-acting GLP1R (glucagon-like peptide-1 receptor) agonist, was recently assessed in Ishikawa EC cells [Citation99]. Liraglutide upregulates the rate of early apoptosis; it arrests Ishikawa cells in the S phase and significantly induces autophagy by increasing LC3 expression and p-PRKAA/AMPKα (Thr172), and by decreasing SQSTM1 protein levels. The combination of liraglutide and 5-aminoimidazole-4-carboximide riboside (AICAR), an AMPK activator, increases the rate of early apoptosis, and enhances autophagosome accumulation, compared with liraglutide alone. This data indicates that liraglutide-induced autophagy might be involved in the induction of cell death. The use of liraglutide to target autophagy could be a potential strategy for EC treatment.
Regarding cellular metabolism, a recent study by Zhou et al. demonstrated that estrogen increases GLS (glutaminase) levels through MYCBP (MYC-binding protein) upregulation, thereby enhancing glutamine metabolism in Ishikawa cells [Citation100]. First, the authors observed that estrogen exposure promotes cell viability and inhibits autophagy in EC cells by decreasing LC3B and BECN1 and increasing SQSTM1 levels and autophagosome and autolysosome structures. Exposing the cells to fulvestrant, an ESR1 inhibitor antagonist, reverses the effects, whereas exposure to glutamine enhances cell viability and inhibits the expression of BECN1, LC3B, and increased SQSTM1. However, treatment with CB-839, a selective oral bioavailable inhibitor of GLS splice variants, negatively regulates glutamine metabolism and inhibits the associated effects of glutamine and estrogen on growth and autophagy both in vitro and in vivo.
Ginsenosides, derivate compounds from ginseng, have demonstrated anti-tumoral activities in several cancers. Protopanaxadiol (PPD), which belongs to the dammarane-type ginsenosides, exerts anti-tumoral activity in EC and acts synergistically with metformin [Citation101]. Since ginsenosides contain a steroidal backbone, they can exert their functions through interactions with steroidal receptors. In fact, PPD plays an antiestrogenic effect and synergizes with tamoxifen in breast cancer cells [Citation102]. In Ishikawa and RL95-2 EC cell lines, PPD and metformin decrease cell viability and induce apoptosis and autophagy by increasing BECN1 protein expression and LC3B-II:LC3B-I ratio. The inhibitory effect of metformin is further increased in combination with PPD. The authors observed that PPD and metformin synergistically regulate the effect of estrogen on Ishikawa and RL95-2 by decreasing the expression of ESR1 (estrogen receptor 1). Combination of PPD and metformin result in decreased tumor growth in Ishikawa cells when subcutaneously injected in mice, compared with both treatments alone.
Progestins
Progesterone therapy is a conservative non-surgical treatment used in patients with early-stage, low-grade, hormone receptor-positive tumors in women who want to maintain their fertility [Citation103]. It is also used as palliative therapy in patients at advanced stages. However, about 30% of these patients are resistant to progesterone treatment [Citation104]. Deciphering the molecular mechanisms of resistance and finding solutions to overcome progesterone resistance represents an urgent clinical need in medical oncology. In this regard, previous studies by Zhuo Z et al. demonstrated that metformin suppresses proliferation, induces cell cycle arrest at G0/G1 and G2/M, and increases apoptosis and autophagy in progesterone-resistant EC cells [Citation105]. This result demonstrates the potential use of metformin in the treatment of progesterone-resistant EC through the modulation of autophagy.
Progestins play key roles in EC treatment as they bind to the progesterone receptor and consequently inhibit cell proliferation and induce autophagy. Liu H et al. reported that long-term exposure to progestin promotes resistance to treatment by activating the PIK3CA-AKT-MTOR signaling pathway, which, in turn, inhibits autophagy [Citation106]. The authors observed that inhibition of the PIK3CA-AKT-MTOR pathway, by means of MTOR knockdown or pharmacological inhibition using RAD001 (everolimus), re-sensitizes progestin-resistant cells to progestins. Similar to other groups [Citation107], the authors generated progestin-resistant cells by exposing EC cells to long-term medroxyprogesterone acetate (MPA) treatment. Importantly, MPA treatment significantly suppresses cell proliferation and cell viability, and induces autophagy in the parental Ishikawa cells but not in progestin-resistant cells, indicating that long-term progestin treatment might decrease progesterone receptor expression levels and activate compensatory pathways to stimulate cell proliferation and inhibit autophagy.
Kinases
Kinases represent a growing area of study in the oncology field. Due to a role in sustaining proliferation and survival of cancer cells, kinases have been reformed into coveted targets for the development of novel anticancer compounds [Citation108,Citation109]. Although kinase inhibitors are considered the major class of novel cancer drugs, it has been described that most tumors can escape single kinase inhibition [Citation109]. Interestingly, in chronic myeloid leukemia and gastrointestinal stromal tumors, which are driven by a single oncogenic kinase, the use of kinase inhibitors presented multi-year increases in survival [Citation110–112]. Survival improvements have also been observed in renal cell carcinoma and other types of cancers that are highly dependent on the angiogenesis process [Citation113–115]. However, in several tumor types like breast, colorectal, lung, pancreatic, prostate, and EC, kinase inhibitors only exhibited modest results [Citation116–122]. In EC, treatments with kinase inhibitors were unsuccessful and tumor progression was characterized by the appearance of refractory cell clones, which are related to increased risk of relapse.
Sorafenib is an FDA-approved multitarget tyrosine and serine/threonine kinase inhibitor, which presents antiangiogenic properties in patients with advanced renal cell carcinoma, hepatocellular carcinoma, and metastatic iodine-resistant thyroid carcinoma [Citation114,Citation123]. Sorafenib has been postulated as a promising targeted treatment for EC given the increased expression levels of VEGFA (vascular endothelial growth factor A) and the prominent role of the RAS-RAF-MAP3K/MEK-MAPK pathway in EC development and progression [Citation124–126]. However, sorafenib presents modest effects in a multi-center phase II clinical trial of advanced uterine carcinoma patients [Citation122].
The mechanisms of sorafenib resistance have been partially elucidated and have the potential to improve the outcome of EC patients [Citation127]. By using bioinformatics analysis and in vitro/in vivo experimental data, Eritja N et al. demonstrated that resistance could be related to the autophagy pathway. Sorafenib treatment results in a dramatic decrease in EIF4E (eukaryotic translation initiation factor 4E) phosphorylated levels and reduced cell viability as measure by cellular DNA loss, increased apoptotic cell numbers, caspase activation and PARP (poly[ADP-ribose] polymerase) cleavage. These results indicate that sorafenib is an effective anticancer drug in vitro, supporting previous results [Citation128]. Nonetheless, before cell commitment to apoptosis takes place, sorafenib exposure triggers an increased LC3B lipidation in Ishikawa, HEC-1A and KLE EC cells, suggesting an increase in autophagosome formation. This data is supported by the increased immunofluorescence of LC3B-II puncta per cell that enhances after autophagy inhibition with CQ. Sorafenib treatment increases SQSTM1 proteolysis, which is reversed by CQ or Baf A1 autophagy inhibitors. Most importantly, a chimeric mRFP-GFP tandem fluorescent-tagged LC3B construct was also used to monitor autophagic flux. Under these conditions, autophagosomes are detected as a yellow signal (merged mRFP and GFP signal). However, under enhanced autophagy, the GFP signal progressively loses fluorescence due to the lysosomal acidic and degradative conditions. The mRFP remains partially stable, thus labeling autolysosomes in red. Accordingly, quantification of red (mRFP+ GFP−) and yellow (mRFP+ GFP+) puncta per cell corroborated that sorafenib induces autophagic flux. Similar results were observed by immunofluorescence, transmitted electron microscopy (TEM), and immunohistochemistry in 3D cultures and in HEC-1A subcutaneous xenografts. Mechanistically, the authors demonstrated that the acute autophagy response stems from early ER stress (XBP1 [X-box binding protein 1] mRNA splicing and increased DDIT3 [DNA damage inducible transcript 3] protein levels) and activation of MAPK8/JNK and JUN. Most importantly, inhibition of MAPK8 (mitogen-activated protein kinase 8) and MAPK9 (mitogen-activated protein kinase 9) by using CEP-1347 or by gene editing impairs the formation of LC3B-II in response to sorafenib. The authors also observed that autophagy inhibition with CQ or BECN1 shRNA sensitizes EC cells to sorafenib, suggesting that sorafenib activates an early protective autophagic response that provides cancer cells with an adaptive response to therapeutic stress. Results were also validated in vivo in subcutaneous HEC-1A tumors and metastatic lung models using MFE-296 EC cells. Authors also generated primary EC orthoxenografts to test the combination of sorafenib-CQ and demonstrated that autophagy inhibition enhances sorafenib cytotoxicity and suppresses tumor growth. In conclusion, sorafenib activates a protective autophagic response in EC cells and the above results highlight a new strategy for therapeutic intervention in recurrent EC.
Another study focused on the function of SGK1 (serum/glucocorticoid regulated kinase 1), a serine/threonine protein kinase that presents homologous function and structure with the AKT family. It is regulated by PDK1 (pyruvate dehydrogenase kinase 1)-mediated phosphorylation [Citation129], by MTORC1 and MTORC2 (mechanistic target of rapamycin kinase complex 2), although more pieces of evidence support the involvement of MTORC2 [Citation130], and the IGF1-INS pathway [Citation131].
SGK1 and SGK3 (serum/glucocorticoid regulated kinase family member 3) are involved in the modulation of cell proliferation and survival in cancer, supporting apoptosis resistance of cancer cells in hostile tumor microenvironments [Citation132]. They are both located downstream of the PI3KCA pathway and interact with the RAS-RAF-ERK pathway and EGFR (epidermal growth factor receptor) [Citation132]. SGK1 is also responsible for stimulating the activity of several ion channels, and its expression is significantly upregulated in EC [Citation133]. A study used an SGK1 inhibitor (SI113) and found that its administration reduces cell viability in Ishikawa, HEC1-B, and AN3CA EC cells [Citation129]. In addition, SI113 treatment induces apoptosis, PARP and CASP9 cleavage, and autophagy through increased LC3B-II and BECN1 expression. SI113 also upregulates endoplasmic reticulum chaperone HSPA5/BiP (heat shock protein family A [HSP70] member 5) and DDIT3 protein levels, suggesting that cell viability after SGK1 inhibition may be compromised by ER stress activation.
Ion channels
Calcium-permeable ion channels and autophagy were reportedly involved in tumor development, progression, and resistance to chemotherapy [Citation134]. As such, intracellular Ca2+ can be regarded as a main regulator of autophagy, and several proteins have been involved in this regulation (e.g., MTORC1 and CAMKK2 [Ca2+/calmodulin-dependent protein kinase kinase 2]) [Citation135]. The role of calcium in autophagy regulation is dependent on a correct spatiotemporal Ca2+ availability with a controlled ionic flow between the extracellular environment, cytosol and storage compartments, growth factors, and nutrient availability, and the type of pathology [Citation134]. The use of nifedipine, an L-type calcium channel antagonist, suppresses proliferation in cancer, and its relation to autophagy, has been studied in EC [Citation136]. Nifedipine inhibits proliferation and migration and increases apoptosis in HEC-1A cells. In addition, treatment with nifedipine significantly induces autophagy by increasing LC3 and BECN1 expression and decreasing RPS6KB1 levels. Treatment with 3-MA in HEC-1A decreases the levels of CACNA1D (calcium voltage-gated channel subunit alpha1 D), evidencing that its protein expression could be positively regulated by autophagy. In addition, inhibition of autophagy with 3-MA increases nifedipine-induced apoptosis and downregulates BECN1 expression, suggesting that autophagy may function as a protective cell survival mechanism. In conclusion, CACNA1D inhibitors (such as nifedipine) could represent potential drug candidates in EC targeted therapy.
Others
Recently, a study by Fukuda et al. described the effect of resveratrol (RSV), a polyphenolic compound derived from red wine, in EC [Citation137]. RSV inhibits proliferation in Ishikawa cells, increases the quantity of the sub G1 population, and activates apoptosis. In addition, RSV exposure induces autophagy, as shown by LC3-II and autophagosome accumulation. Treatment with both RSV and CQ or knockdown of ATG5 or ATG7 increases RSV-induced apoptosis significantly, suggesting that RSV-induced autophagy might counteract the antitumor effect of RSV in Ishikawa cells.
Similarly, Wu et al. reported the effects of isoliquiritigenin (ISL), a natural flavonoid obtained from the root of licorice (Glycyrrhiza uralensis) with a chalcone structure (4, 20, 40-trihydroxychalcone). In addition to its anti-oxidant, anti-inflammatory, and anti-platelet aggregation properties [Citation138–140], ISL has also been described as an anti-tumoral, inhibitor of cell growth and apoptosis inducer [Citation141]. As such, ISL induces cell cycle arrest at the sub G1 and G2/M phases in Ishikawa and HEC-1A cells, respectively, and apoptosis by increasing the levels of cleaved CASP3, CASP7 (caspase 7), and PARP [Citation142]. The treatment with ISL also activates the DNA damage marker H2AX (H2A.X variant histone) and increases TP53 phosphorylation and CDKN1A levels in both cell lines. In addition, ISL induces autophagy, as shown by increased autophagosomes, LC3-II levels, and SQSTM1 expression. The authors also reported that ERK signaling is necessary for ISL-induced apoptosis and autophagy. Combination of ISL and Baf A1 slightly increases ISL-mediated cell cycle arrest and apoptosis in HEC-1A, suggesting that ISL-induced autophagy plays a cell survival protective role in tumor cells. Finally, ISL treatment of HEC-1A subcutaneous xenografts dramatically inhibits tumor growth when compared to vehicle control.
Considerations and remarks
Despite an increase in studies exploring the role of autophagy in the endometrium, a significant number of reports are exclusively based on histological expression analysis or overly dependent on the use of pharmacological agents such as chloroquine (CQ), 3-methyladenine (3-MA), or bafilomycin A1. While this information helps decipher the role of autophagy in the endometrium, the functional conclusions and insights that can be extracted are far more limited. This is due, for instance, to the limited assurances that these approaches can offer to accurately analyze and reliably monitor autophagic flux increased on-rates or decreased off-rates in contrast to other state-of-the-art methodologies (e.g., tandem GFP-LC3-RFP based techniques) [Citation3,Citation6]. Unfortunately, a considerable number of studies in EC commonly use static autophagy techniques (e.g., electron microscopy or LC3-II immune detection). Although regarded as valuable complementary information, these techniques are generally considered unsuitable for this purpose. Similarly, the same can be contextualized when performing in vivo analyses. For instance, pending on the evaluation of probe expression and time resolution, to the best of our knowledge the monitoring and quantitation of autophagic flux in the endometrium has not yet been addressed using genetically modified mouse models suitable for this purpose, such as the mRFP-GFP-LC3 [Citation143] or GFP-LC3-RFP-LC3∆G [Citation144] reporter mice.
Finally, another important question relates to the use of chemical agents and strategies designed to target autophagy, mainly 3-MA and CQ. Repurposing of CQ derivatives, such as hydroxychloroquine, have reached encouraging results in phase-II clinical trials [Citation145]. However, limitations in terms of specificity, potency, and cell penetration in acidic microenvironments have prevented final approval as a treatment option. Consequently, the therapeutic potential of autophagy modulation has not been fully exploited. Additional efforts are being conducted to develop novel compounds with improved specificity and potency. Currently, there is a focus on compound inhibitors targeting PIK3C3/Vps34 (phosphatidylinositol 3-kinase catalytic subunit type 3) [Citation146–149], ULK1 [Citation150,Citation151] or the PIKFYVE (phosphoinositide kinase, FYVE-type zinc finger containing) [Citation152,Citation153]. These compounds showed promising preclinical efficacy against multiple cancer cell types, yet none of them have been previously tested on EC cells. Importantly, the use of these compounds, or the design of new ones, to study autophagy in the endometrium may provide the scientific community with unanticipated results in the field of EC. Other compounds used for inhibiting or increasing autophagy are reviewed in detail elsewhere [Citation6]. In conclusion, more rigorous assessment of the autophagic flux, as well as additional mechanistic studies, are necessary to understand the role that autophagy plays in the endometrium and to ultimately transfer this knowledge into the clinical setting by designing new drugs.
Conclusions
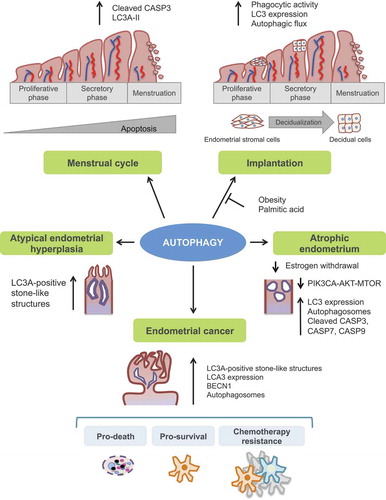
Currently available data indicates that autophagy represents a pivotal cellular process that is modulated cyclically during the menstrual cycle. Autophagy has important implications in normal uterine function, reproductive biology, and endometrial atrophy. An increasingly large body of evidence indicates that autophagy also plays an important role in the development of endometrial cancer (). In this regard, autophagy impacts endometrial cancer response to standard chemotherapy/radiotherapy and targeted agents with potential implications in the immune system, given its immune-modulatory properties. Additional studies and detailed mechanistic insights are required to fully elucidate and decipher the function of autophagy in the endometrium. Improved standardized protocols are also necessary to solve some of the paradoxical conclusions that can be drawn from the above studies, especially regarding the context-dependent roles of autophagy.
Figure 3. Illustration of the principal autophagy-related function into the human endometrium. Autophagy has a crucial role in both normal and pathological endometrium and has been linked to endometrial cancer development
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Farré J-C, Subramani S. Mechanistic insights into selective autophagy pathways: lessons from yeast. Nat Rev Mol Cell Biol. 2016;17:537–552.
- Ryter SW, Cloonan SM, Choi AMK. Autophagy: a critical regulator of cellular metabolism and homeostasis. Mol Cells. 2013;36:7–16.
- Klionsky DJ, Abdelmohsen K, Abe A, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy. 2016;12:1–222.
- Yu L, Chen Y, Tooze SA. Autophagy pathway: cellular and molecular mechanisms. Autophagy. 2018;14:207–215.
- Galluzzi L, Baehrecke EH, Ballabio A, et al. Molecular definitions of autophagy and related processes. Embo J. 2017;36:1811–1836.
- Galluzzi L, Bravo-San Pedro JM, Levine B, et al. Pharmacological modulation of autophagy: therapeutic potential and persisting obstacles. Nat Rev Drug Discov. 2017;16:487–511.
- Cuervo AM, Wong E. Chaperone-mediated autophagy: roles in disease and aging. Cell Res. 2014;24:92–104.
- Kaushik S, Cuervo AM. Chaperone-mediated autophagy: a unique way to enter the lysosome world. Trends Cell Biol. 2012;22:407–417.
- Mijaljica D, Prescott M, Devenish RJ. Microautophagy in mammalian cells: revisiting a 40-year-old conundrum. Autophagy. 2011;7:673–682.
- Mizushima N, Yoshimori T, Ohsumi Y. The role of atg proteins in autophagosome formation. Annu Rev Cell Dev Biol. 2011;27:107–132.
- Noda NN, Inagaki F. Mechanisms of autophagy. Annu Rev Biophys. 2015;44:101–122.
- Levine B, Klionsky DJ. Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev Cell. 2004;6:463–477.
- Lamb CA, Yoshimori T, Tooze SA. The autophagosome: origins unknown, biogenesis complex. Nat Rev Mol Cell Biol. 2013;14:759–774.
- Green DR, Galluzzi L, Kroemer G. Mitochondria and the autophagy-inflammation-cell death axis in organismal aging. Science. 2011;333:1109–1112.
- Mizushima N, Levine B. Autophagy in mammalian development and differentiation. Nat Cell Biol. 2010;12:823–830.
- Beier HM, Beier-Hellwig K. Molecular and cellular aspects of endometrial receptivity. Hum Reprod Update. 1998;4:448–458.
- Ruiz-Alonso M, Blesa D, Simón C. The genomics of the human endometrium. Biochim Biophys Acta - Mol Basis Dis. 2012;1822:1931–1942.
- Talbi S, Hamilton AE, Vo KC, et al. Molecular phenotyping of human endometrium distinguishes menstrual cycle phases and underlying biological processes in normo-ovulatory women. Endocrinology. 2006;147:1097–1121.
- Harada T, Kaponis A, Iwabe T, et al. Apoptosis in human endometrium and endometriosis. Hum Reprod Update. 2004;10:29–38.
- Bulun S, Adashi E. The Physiology and Pathology of the Female Reproductive Axis. In: Kronenberg HM, Melmed S, Polonsky KS, et al., editors. Williams textbook of endocrinology. Philadelphia, PA:Elsevier Health Sciences; 2007. p. 587–663.
- Choi J, Jo M, Lee E, et al. The role of autophagy in human endometrium. Biol Reprod. 2012;86:70.
- Cakmak H, Taylor HS. Implantation failure: molecular mechanisms and clinical treatment. Hum Reprod Update. 2011;17:242–253.
- Su R-W, Fazleabas AT. Implantation and establishment of pregnancy in human and nonhuman primates. Adv Anat Embryol Cell Biol. 2015;216:189–213.
- Kim S-M, Kim J-S. A review of mechanisms of implantation. Dev Reprod. 2017;21:351–359.
- Cornillie FJ, Lauweryns JM, Brosens IA. Normal human Endometrium. Gynecol Obstet Invest. 1985;20:113–129.
- Lawn AM, Wilson EW, Finn CA. The ultrastructure of human decidual and predecidual cells. J Reprod Fertil. 1971;26:85–90.
- Avagliano L, Terraneo L, Virgili E, et al. Autophagy in normal and abnormal early human pregnancies. Reprod Sci. 2015;22:838–844.
- Mestre Citrinovitz AC, Strowitzki T, Germeyer A. Decreased autophagy impairs decidualization of human endometrial stromal cells: a role for ATG proteins in endometrial physiology. Int J Mol Sci. 2019;20:3066.
- Rhee JS, Saben JL, Mayer AL, et al. Diet-induced obesity impairs endometrial stromal cell decidualization: a potential role for impaired autophagy. Hum Reprod. 2016;31:1315–1326.
- Talmor A, Dunphy B. Female obesity and infertility. Best Pract Res Clin Obstet Gynaecol. 2015;29:498–506.
- Boots CE, Bernardi LA, Stephenson MD. Frequency of euploid miscarriage is increased in obese women with recurrent early pregnancy loss. Fertil Steril. 2014;102:455–459.
- Luzzo KM, Wang Q, Purcell SH, et al. High fat diet induced developmental defects in the mouse: oocyte meiotic aneuploidy and fetal growth retardation/brain defects. PLoS One. 2012;7:e49217.
- Nakashima A, Aoki A, Kusabiraki T, et al. Role of autophagy in oocytogenesis, embryogenesis, implantation, and pathophysiology of pre-eclampsia. J Obstet Gynaecol Res. 2017;43:633–643.
- Zhou S, Zhao L, Yi T, et al. Menopause-induced uterine epithelium atrophy results from arachidonic acid/prostaglandin E2 axis inhibition-mediated autophagic cell death. Sci Rep. 2016;6:31408.
- Zhang D, Li J, Xu G, et al. Follicle-stimulating hormone promotes age-related endometrial atrophy through cross-talk with transforming growth factor beta signal transduction pathway. Aging Cell. 2015;14:284–287.
- Santoro N, Randolph JF. Reproductive hormones and the menopause transition. Obstet Gynecol Clin North Am. 2011;38:455–466.
- Sanderson PA, Critchley HOD, Williams ARW, et al. New concepts for an old problem: the diagnosis of endometrial hyperplasia. Hum Reprod Update. 2017;23:232–254.
- Baak JPA, Mutter GL. EIN and WHO94. J Clin Pathol. 2005;58:1–6.
- Mutter GL. Endometrial intraepithelial Neoplasia (EIN): will it bring order to Chaos? Gynecol Oncol. 2000;76:287–290.
- Jarboe EA, Mutter GL. Endometrial intraepithelial neoplasia. Semin Diagn Pathol. 2010;27:215–225.
- Feng L, Li J, Yang L, et al. Tamoxifen activates Nrf2-dependent SQSTM1 transcription to promote endometrial hyperplasia. Theranostics. 2017;7:1890–1900.
- Strissel PL, Ellmann S, Loprich E, et al. Early aberrant insulin-like growth factor signaling in the progression to endometrial carcinoma is augmented by tamoxifen. Int J Cancer. 2008;123:2871–2879.
- Lin BC, Suzawa M, Blind RD, et al. Stimulating the GPR30 estrogen receptor with a novel tamoxifen analogue activates SF-1 and promotes endometrial cell proliferation. Cancer Res. 2009;69:5415–5423.
- Sivridis E, Giatromanolaki A, Liberis V, et al. Autophagy in endometrial carcinomas and prognostic relevance of “stone-like” structures (SLS): what is destined for the atypical endometrial hyperplasia? Autophagy. 2011;7:74–82.
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 2018;68:7–30.
- Ferlay J, Soerjomataram I, Dikshit R, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136(5):E359–86.
- Sorosky JI. Endometrial cancer. Obstet Gynecol. 2012;120(2, Part 1):383–397.
- Bokhman JV. Two pathogenetic types of endometrial carcinoma. Gynecol Oncol. 1983;15(1):10–17.
- Matias-Guiu X, Prat J. Molecular pathology of endometrial carcinoma. Histopathology. 2013;62(1):111–123.
- Llobet D, Pallares J, Yeramian A, et al. Molecular pathology of endometrial carcinoma: practical aspects from the diagnostic and therapeutic viewpoints. J Clin Pathol. 2009;62(9):777–785.
- Matias-Guiu X, Davidson B. Prognostic biomarkers in endometrial and ovarian carcinoma. Virchows Arch. 2014;464(3):315–331.
- Levine DA, Getz G, Gabriel S, et al. Integrated genomic characterization of endometrial carcinoma. Nature. 2013;497(7447):67–73.
- Lebovitz CB, Robertson AG, Goya R, et al. Cross-cancer profiling of molecular alterations within the human autophagy interaction network. Autophagy. 2015;11(9):1668–1687.
- Budina-Kolomets A, Hontz RD, Pimkina J, et al. A conserved domain in exon 2 coding for the human and murine ARF tumor suppressor protein is required for autophagy induction. Autophagy. 2013;9(10):1553–1565.
- Yemelyanova A, Ji H, Shih I-M, et al. Utility of p16 expression for distinction of uterine serous carcinomas from endometrial endometrioid and endocervical adenocarcinomas. Am J Surg Pathol. 2009;33(10):1504–1514.
- Giatromanolaki A, Koukourakis MI, Koutsopoulos A, et al. High Beclin 1 expression defines a poor prognosis in endometrial adenocarcinomas. Gynecol Oncol. 2011;123(1):147–151.
- Zhao J-H, Wan X-Y, Xie X, et al. Expression and clinical significance of Beclin1 and PTEN in endometrial carcinoma. Ai Zheng Internet. 2006;25:753–757.
- Orfanelli T, Jeong JM, Doulaveris G, et al. Involvement of autophagy in cervical, endometrial and ovarian cancer. Int J Cancer. 2014;135(3):519–528.
- Deng L. Identification of a novel estrogen-regulated gene, EIG121, induced by hormone replacement therapy and differentially expressed in type I and Type II endometrial cancer. Clin Cancer Res. 2005;11(23):8258–8264.
- Deng L, Feng J, Broaddus RR. The novel estrogen-induced gene EIG121 regulates autophagy and promotes cell survival under stress. Cell Death Dis. 2010;1(4):e32–e32.
- Nout RA, Smit VTHBM, Putter H, et al. Vaginal brachytherapy versus pelvic external beam radiotherapy for patients with endometrial cancer of high-intermediate risk (PORTEC-2): an open-label, non-inferiority, randomised trial. Lancet. 2010;375(9717):816–823.
- Blake P, Swart AM, Orton J, et al.; ASTEC/EN.5 Study Group. Adjuvant external beam radiotherapy in the treatment of endometrial cancer (MRC ASTEC and NCIC CTG EN.5 randomised trials): pooled trial results, systematic review, and meta-analysis. Lancet. 2009;373:137–146.
- Tangjitgamol S, See HT, Kavanagh J. Adjuvant chemotherapy for endometrial cancer. Int J Gynecol Cancer. 2011;21(5):885–895.
- Fleming GF. Systemic chemotherapy for uterine carcinoma: metastatic and adjuvant. J Clin Oncol. 2007;25(20):2983–2990.
- Eritja N, Yeramian A, Chen B-J, et al. Endometrial carcinoma: specific targeted pathways. Adv Exp Med Biol. 2017;943:149–207.
- Santaballa A, Matías-Guiu X, Redondo A, et al. SEOM clinical guidelines for endometrial cancer (2017). Clin Transl Oncol. 2018;20(1):29–37.
- Morselli E, Galluzzi L, Kepp O, et al. Anti- and pro-tumor functions of autophagy. Biochim Biophys Acta Mol Cell Res. 2009;1793(9):1524–1532.
- Mohammad RM, Muqbil I, Lowe L, et al. Broad targeting of resistance to apoptosis in cancer. Semin Cancer Biol. 2015;35:S78–103.
- Notte A, Leclere L, Michiels C. Autophagy as a mediator of chemotherapy-induced cell death in cancer. Biochem Pharmacol. 2011;82(5):427–434.
- Shintani T. Autophagy in health and disease: a double-edged sword. Science. 2004;306(5698):990–995.
- Fukuda T, Oda K, Wada-Hiraike O, et al. The anti-malarial chloroquine suppresses proliferation and overcomes cisplatin resistance of endometrial cancer cells via autophagy inhibition. Gynecol Oncol. 2015;137(3):538–545.
- Sun M-Y, Zhu J-Y, Zhang C-Y, et al. Autophagy regulated by lncRNA HOTAIR contributes to the cisplatin-induced resistance in endometrial cancer cells. Biotechnol Lett. 2017;39(10):1477–1484.
- Lin Q, Wang Y, Chen D, et al. Cisplatin regulates cell autophagy in endometrial cancer cells via the PI3K/AKT/mTOR signalling pathway. Oncol Lett. 2017;13(5):3567–3571.
- Richardson PG, Anderson KC. Bortezomib: a novel therapy approved for multiple myeloma. Clin Adv Hematol Oncol. 2003;1(10):596–600.
- Dolcet X, Llobet D, Encinas M, et al. Proteasome inhibitors induce death but activate NF-κB on endometrial carcinoma cell lines and primary culture explants. J Biol Chem. 2006;281(31):22118–22130.
- Periyasamy-Thandavan S, Jackson WH, Samaddar JS, et al. Bortezomib blocks the catabolic process of autophagy via a cathepsin-dependent mechanism, affects endoplasmic reticulum stress, and induces caspase-dependent cell death in antiestrogen–sensitive and resistant ER+ breast cancer cells. Autophagy. 2010;6(1):19–35.
- Hong YS, Hong S-W, Kim S-M, et al. Bortezomib induces G2-M arrest in human colon cancer cells through ROS-inducible phosphorylation of ATM-CHK1. Int J Oncol. 2012;41(1):76–82. .
- Befani CD, Vlachostergios PJ, Hatzidaki E, et al. Bortezomib represses HIF-1α protein expression and nuclear accumulation by inhibiting both PI3K/Akt/TOR and MAPK pathways in prostate cancer cells. J Mol Med. 2012;90(1):45–54.
- Chen D, Frezza M, Schmitt S, et al. Bortezomib as the first proteasome inhibitor anticancer drug: current status and future perspectives. Curr Cancer Drug Targets. 2011;11(3):239–253.
- Kao C, Chao A, Tsai C-L, et al. Bortezomib enhances cancer cell death by blocking the autophagic flux through stimulating ERK phosphorylation. Cell Death Dis. 2014;5(11):e1510–e1510.
- Eckschlager T, Plch J, Stiborova M, et al. Histone deacetylase inhibitors as anticancer drugs. Int J Mol Sci. 2017;18(7):1414.
- De Umasankar U, Son J, Sachan R, et al. A new synthetic histone deacetylase inhibitor, MHY2256, induces apoptosis and autophagy cell death in endometrial cancer cells via p53 acetylation. Int J Mol Sci. 2018;19(9):2743.
- Zhou W-J, Chang -K-K, Wu K, et al. Rapamycin synergizes with cisplatin in antiendometrial cancer activation by improving IL-27–stimulated cytotoxicity of NK cells. Neoplasia. 2018;20(1):69–79.
- Zingoni A, Fionda C, Borrelli C, et al. Natural killer cell response to chemotherapy-stressed cancer cells: role in tumor immunosurveillance. Front Immunol. 2017;8:1194.
- Vandenput I, Vergote I, Leunen K, et al. Leuven dose-dense paclitaxel/carboplatin regimen in patients with primary advanced or recurrent endometrial carcinoma. Int J Gynecol Cancer. 2009;19(6):1147–1151.
- Sorbe B, Andersson H, Boman K, et al. Treatment of primary advanced and recurrent endometrial carcinoma with a combination of carboplatin and paclitaxellong-term follow-up. Int J Gynecol Cancer. 2008;18(4):803–808.
- Felip I, Moiola CP, Megino-Luque C, et al. Therapeutic potential of the new TRIB3-mediated cell autophagy anticancer drug ABTL0812 in endometrial cancer. Gynecol Oncol. 2019;153(2):425–435. .
- Liu S, Li X. Autophagy inhibition enhances sensitivity of endometrial carcinoma cells to paclitaxel. Int J Oncol. 2015;46(6):2399–2408.
- Ran X, Yang J, Liu C, et al. MiR-218 inhibits HMGB1-mediated autophagy in endometrial carcinoma cells during chemotherapy. Int J Clin Exp Pathol. 2015;8(6):6617–6626.
- Wang H, Li D, Li X, et al. Mammalian target of rapamycin inhibitor RAD001 sensitizes endometrial cancer cells to paclitaxel-induced apoptosis via the induction of autophagy. Oncol Lett. 2016;12(6):5029–5035.
- Ran X, Zhou P, Zhang K. Autophagy plays an important role in stemness mediation and the novel dual function of EIG121 in both autophagy and stemness regulation of endometrial carcinoma JEC cells. Int J Oncol. 2017;51(2):644–656.
- Yoshida GJ. Metabolic reprogramming: the emerging concept and associated therapeutic strategies. J Exp Clin Cancer Res. 2015;34(1):111.
- Kasznicki J, Sliwinska A, Drzewoski J. Metformin in cancer prevention and therapy. Ann Transl Med. 2014;2:57.
- Quinn BJ, Kitagawa H, Memmott RM, et al. Repositioning metformin for cancer prevention and treatment. Trends Endocrinol Metab. 2013;24(9):469–480.
- Meng S, Cao J, He Q, et al. Metformin activates AMP-activated protein kinase by promoting formation of the αβγ heterotrimeric complex. J Biol Chem. 2015;290(6):3793–3802.
- Ben Sahra I, Regazzetti C, Robert G, et al. Metformin, Independent of AMPK, induces mTOR inhibition and cell-cycle arrest through REDD1. Cancer Res. 2011;71(13):4366–4372.
- Takahashi A, Kimura F, Yamanaka A, et al. Metformin impairs growth of endometrial cancer cells via cell cycle arrest and concomitant autophagy and apoptosis. Cancer Cell Int. 2014;14(1):53.
- Nadkarni P, Chepurny OG, Holz GG. Regulation of glucose homeostasis by GLP-1. Prog Mol Biol Transl Sci. 2014;121:23–65.
- Kanda R, Hiraike H, Wada-Hiraike O, et al. Expression of the glucagon-like peptide-1 receptor and its role in regulating autophagy in endometrial cancer. BMC Cancer. 2018;18(1):657.
- Zhou W-J, Zhang J, Yang H-L, et al. Estrogen inhibits autophagy and promotes growth of endometrial cancer by promoting glutamine metabolism. Cell Commun Signal. 2019;17(1):99.
- Gu C-J, Cheng J, Zhang B, et al. Protopanaxadiol and metformin synergistically inhibit estrogen-mediated proliferation and anti-autophagy effects in endometrial cancer cells. Am J Transl Res. 2017;9(9):4071–4082.
- Yu Y, Zhou Q, Hang Y, et al. Antiestrogenic effect of 20S-protopanaxadiol and its synergy with tamoxifen on breast cancer cells. Cancer. 2007;109(11):2374–2382.
- Kim J, Chapman-Davis E. Role of progesterone in endometrial cancer. Seminars in Reproductive Medicine. 2010;28(1):081–90.
- Dizon DS. Treatment options for advanced endometrial carcinoma. Gynecol Oncol. 2010;117(2):373–381.
- Zhuo Z, Wang A, Yu H. Metformin targeting autophagy overcomes progesterone resistance in endometrial carcinoma. Arch Gynecol Obstet. 2016;294(5):1055–1061.
- Liu H, Zhang L, Zhang X, et al. PI3K/AKT/mTOR pathway promotes progestin resistance in endometrial cancer cells by inhibition of autophagy. Onco Targets Ther. 2017;10:2865–2871.
- Zhao S, Li G, Yang L, et al. Response-specific progestin resistance in a newly characterized Ishikawa human endometrial cancer subcell line resulting from long-term exposure to medroxyprogesterone acetate. Oncol Lett. 2013;5(1):139–144.
- Gschwind A, Fischer OM, Ullrich A. The discovery of receptor tyrosine kinases: targets for cancer therapy. Nat Rev Cancer. 2004;4(5):361–370.
- Knight ZA, Lin H, Shokat KM. Targeting the cancer kinome through polypharmacology. Nat Rev Cancer. 2010;10(2):130–137.
- Druker BJ, Guilhot F, O’Brien SG, et al. Five-year follow-up of patients receiving imatinib for chronic myeloid leukemia. N Engl J Med. 2006;355(23):2408–2417.
- Heinrich MC, Corless CL, Demetri GD, et al. Kinase mutations and imatinib response in patients with metastatic gastrointestinal stromal tumor. J Clin Oncol. 2003;21(23):4342–4349.
- Druker BJ, Talpaz M, Resta DJ, et al. Efficacy and Safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N Engl J Med. 2001;344(14):1031–1037.
- Motzer RJ, Hutson TE, Tomczak P, et al. Sunitinib versus interferon Alfa in metastatic renal-cell carcinoma. N Engl J Med. 2012;22(2):407–417.
- Escudier B, Eisen T, Stadler WM, et al. Sorafenib in advanced clear-cell renal-cell carcinoma. N Engl J Med. 2007;356:125–134.
- Yang JC, Haworth L, Sherry RM, et al. A randomized trial of bevacizumab, an anti–vascular endothelial growth factor antibody, for metastatic renal cancer. N Engl J Med. 2003;349:427–434.
- Shepherd FA, Rodrigues Pereira J, Ciuleanu T, et al. Erlotinib in previously treated non–small-cell lung cancer. N Engl J Med. 2011;333(2):1109–1112.
- Sandler A, Gray R, Perry MC, et al. Paclitaxel–carboplatin alone or with bevacizumab for non–small-cell lung cancer. N Engl J Med. 2006;355(24):2542–2550.
- Geyer CE, Forster J, Lindquist D, et al. Lapatinib plus capecitabine for HER2-positive advanced breast cancer. N Engl J Med. 2004;10(26):29–38.
- Miller K, Wang M, Gralow J, et al. Paclitaxel plus bevacizumab versus paclitaxel alone for metastatic breast cancer. N Engl J Med. 2007;357(26):2666–2676.
- Jonker DJ, O’Callaghan CJ, Karapetis CS, et al. Cetuximab for the treatment of colorectal cancer. N Engl J Med. 2007;357(20):2040–2048.
- Moore MJ, Goldstein D, Hamm J, et al. Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: a phase III trial of the national cancer institute of Canada clinical trials group. J Clin Oncol. 2007;25(15):1960–1966. .
- Nimeiri HS, Oza AM, Morgan RJ, et al. A phase II study of sorafenib in advanced uterine carcinoma/carcinosarcoma: A trial of the Chicago, PMH, and California Phase II Consortia. Gynecol Oncol. 2010;117(1):37–40.
- Bruix J, Sherman M. American association for the study of liver diseases. management of hepatocellular carcinoma: an update. Hepatology. 2011;53(3):1020–1022.
- Kamat AA, Merritt WM, Coffey D, et al. Clinical and biological significance of vascular endothelial growth factor in endometrial cancer. Clin Cancer Res. 2007;13(24):7487–7495.
- McMeekin DS, Sill MW, Darcy KM, et al. A phase II trial of thalidomide in patients with refractory leiomyosarcoma of the uterus and correlation with biomarkers of angiogenesis: A gynecologic oncology group study. Gynecol Oncol. 2007;106(3):596–603.
- Ninomiya Y. K-Ras and H-Ras activation promote distinct consequences on endometrial cell survival. Cancer Res. 2004;64(8):2759–2765.
- Eritja N, Chen B-J, Rodríguez-Barrueco R, et al. Autophagy orchestrates adaptive responses to targeted therapy in endometrial cancer. Autophagy. 2017;13(3):608–624.
- Llobet D, Eritja N, Yeramian A, et al. The multikinase inhibitor Sorafenib induces apoptosis and sensitises endometrial cancer cells to TRAIL by different mechanisms. Eur J Cancer. 2010;46(4):836–850.
- Conza D, Mirra P, Calì G, et al. The SGK1 inhibitor SI113 induces autophagy, apoptosis, and endoplasmic reticulum stress in endometrial cancer cells. J Cell Physiol. 2017;232:3735–3743.
- García-Martínez JM, Alessi DR. mTOR complex 2 (mTORC2) controls hydrophobic motif phosphorylation and activation of serum- and glucocorticoid-induced protein kinase 1 (SGK1). Biochem J. 1985;20(3):113–129.
- Perrotti N, He RA, Phillips SA, et al. Activation of serum- and glucocorticoid-induced protein kinase (Sgk) by Cyclic AMP and Insulin. J Biol Chem. 2019;20(12):9406–9412.
- Bruhn MA, Pearson RB, Hannan RD, et al. Second AKT: the rise of SGK in cancer signalling. Growth Factors. 2010;28(6):394–408.
- Lang F, Shumilina E. Regulation of ion channels by the serum- and glucocorticoid-inducible kinase SGK1. Faseb J. 2015;14(1):3–12.
- Kondratskyi A. Calcium-permeable ion channels in control of autophagy and cancer. Front Physiol. 2010;27:272.
- Filippi-Chiela EC, Viegas MS, Thome MP, et al. Modulation of autophagy by calcium signalosome in human disease. Mol Pharmacol. 2018;68(3):7–30.
- Bao -X-X, Xie B-S, Li Q, et al. Nifedipine induced autophagy through Beclin1 and mTOR pathway in endometrial carcinoma cells. Chin Med J. 2014;464(17):315–331.
- Fukuda T, Oda K, Wada-Hiraike O, et al. The novel estrogen-induced gene EIG121 regulates autophagy and promotes cell survival under stress. Oncol Lett. 2007;25(4):2983–2990. .
- Haraguchi H, Ishikawa H, Mizutani K, et al. Antioxidative and superoxide scavenging activities of retrochalcones in Glycyrrhiza inflata. Bioorg Med Chem. 1998;6:339–347.
- Tawata M, Aida K, Noguchi T, et al. Anti-platelet action of isoliquiritigenin, an aldose reductase inhibitor in licorice. Eur J Pharmacol. 2008;18(1):87–92.
- Tamir S, Eizenberg M, Somjen D, et al. Estrogen-like activity of glabrene and other constituents isolated from licorice root. J Steroid Biochem Mol Biol. 2018;18(3):291–298.
- Li T, Satomi Y, Katoh D, et al. Induction of cell cycle arrest and p21CIP1/WAF1 expression in human lung cancer cells by isoliquiritigenin. Cancer Lett. 2004;207:27–35.
- Wu C-H, Chen H-Y, Wang C-W, et al. Isoliquiritigenin induces apoptosis and autophagy and inhibits endometrial cancer growth in mice. Oncotarget. 2016;7(45):73432–73447.
- Li L, Wang ZV, Hill JA, et al. New autophagy reporter mice reveal dynamics of proximal tubular autophagy. J Am Soc Nephrol. 2001;276(2):9406–9412.
- Kaizuka T, Morishita H, Hama Y, et al. An autophagic flux probe that releases an internal control. Mol Cell. 2016;64:835–849.
- Amaravadi RK, Kimmelman AC, Debnath J. Targeting autophagy in cancer: recent advances and future directions. Cancer Discov. 2019;9:1167–1181.
- Pasquier B. SAR405, a PIK3C3/Vps34 inhibitor that prevents autophagy and synergizes with MTOR inhibition in tumor cells. Autophagy. 2015;11:725–726.
- Ronan B, Flamand O, Vescovi L, et al. A highly potent and selective Vps34 inhibitor alters vesicle trafficking and autophagy. Nat Chem Biol. 2014;10:1013–1019.
- Pasquier B, El-Ahmad Y, Filoche-Rommé B, et al. Discovery of (2 S)-8-[(3 R)-3-Methylmorpholin-4-yl]-1-(3-methyl-2-oxobutyl)-2-(trifluoromethyl)-3,4-dihydro-2 H -pyrimido[1,2- a]pyrimidin-6-one: A Novel Potent and Selective Inhibitor of Vps34 for the treatment of solid tumors. J Med Chem. 2015;58:376–400.
- Dyczynski M, Yu Y, Otrocka M, et al. Targeting autophagy by small molecule inhibitors of vacuolar protein sorting 34 (Vps34) improves the sensitivity of breast cancer cells to Sunitinib. Cancer Lett. 2018;435:32–43.
- Egan DF, Chun MGH, Vamos M, et al. Small molecule inhibition of the autophagy kinase ULK1 and identification of ULK1 substrates. Mol Cell. 2015;59:285–297.
- Martin KR, Celano SL, Solitro AR, et al. A potent and selective ULK1 inhibitor suppresses autophagy and sensitizes cancer cells to nutrient stress. iScience. 2018;8:74–84.
- Sharma G, Guardia CM, Roy A, et al. A family of PIKFYVE inhibitors with therapeutic potential against autophagy-dependent cancer cells disrupt multiple events in lysosome homeostasis. Autophagy. 2019;15:1694–1718.
- Gayle S, Landrette S, Beeharry N, et al. Identification of apilimod as a first-in-class PIKfyve kinase inhibitor for treatment of B-cell non-Hodgkin lymphoma. Blood. 2017;129:1768–1778.