
ABSTRACT
Coxsackievirus B3 (CVB3) is a prevalent etiological agent for viral myocarditis and neurological disorders, particularly in infants and young children. Virus-encoded proteinases have emerged as cytopathic factors that contribute to disease pathogenesis in part through targeting the cellular recycling machinery of autophagy. Although it is appreciated that CVB3 can usurp cellular macroautophagy/autophagy for pro-viral functions, the precise mechanisms by which viral proteinases disrupt autophagy remain incompletely understood. Here we identified TFEB (transcription factor EB), a master regulator of autophagy and lysosome biogenesis, as a novel target of CVB3 proteinase 3 C. Time-course infections uncovered a significant loss of full-length TFEB and the emergence of a lower-molecular mass (~63 kDa) fragment. Cellular and in vitro cleavage assays revealed the involvement of viral proteinase 3 C in the proteolytic processing of TFEB, while site-directed mutagenesis identified the site of cleavage after glutamine 60. Assessment of TFEB transcriptional activity using a reporter construct discovered a loss of function of the cleavage fragment despite nuclear localization and retaining of the ability of DNA and protein binding. Furthermore, we showed that CVB3 infection was also able to trigger cleavage-independent nuclear translocation of TFEB that relied on the serine-threonine phosphatase PPP3/calcineurin. Finally, we demonstrated that both TFEB and TFEB [Δ60] serve roles in viral egress albeit through differing mechanisms. Collectively, this study reveals that CVB3 targets TFEB for proteolytic processing to disrupt host lysosomal function and enhance viral infection.
Abbreviations:ACTB: actin beta; CLEAR: coordinated lysosomal enhancement and regulation; CVB3: coxsackievirus B3; DAPI: 4′,6-diamidino-2-phenylindole; GFP: green fluorescent protein; LAMP1: lysosomal associated membrane protein 1; LTR: LysoTracker Red; PPP3/calcineurin: protein phosphatase 3; PPP3CA: protein phosphatase 3 catalytic subunit A; p-TFEB: phospho-Ser211 TFEB; si-CON: scramble control siRNA; TFEB: transcription factor EB; TFEB [Δ60]: TFEB cleavage fragment that lacks the first 60 amino acids; VP1: viral capsid protein 1
Introduction
Enteroviruses (EVs) are among the most common human pathogens, responsible for an estimated ~1 billion global infections annually [Citation1]. Although infections are subclinical in more than 90% of individuals, EV infection of infants, toddlers, and adolescents can manifest in various illnesses ranging in severity from febrile ailment and neurological disorders (e.g., aseptic meningitis, encephalitis, and non-polio flaccid paralysis) to much severe outcomes, such as viral myocarditis-induced sudden death in young adults. Non-polio EVs such as EV-A71, EV-D68 and coxsackievirus B3 (CVB3) continue to be a global health challenge due to an absence of effective anti-enteroviral therapy [Citation2].
Efforts to understand the pathogenesis of EVs have revealed troves of mechanistic insights into viral subversion of the cellular machineries. In particular, EVs have evolved various strategies to hijack autophagy, an evolutionarily conserved cellular recycling process that normally functions to clear invading pathogens [Citation3]. Specialized membranes termed phagophores first enwrap cytosolic wastes in the form of protein aggregates, damaged organelles, and invading microbes. Upon closure, the double membrane vesicles, so-called autophagosomes, are subsequently trafficked to lysosomes, the major digestive organelles of the cell that maintain a hydrolase-enriched acidic lumen. Following autophagosome fusion with lysosomes, the enwrapped cargo, including the inner membrane of the autophagosomes, is degraded by hydrolysis. Traditionally believed to be only responsible for cellular waste disposal, lysosomes have emerged as versatile signaling hubs that regulate nutrient sensing, transcriptional activity, and autophagy [Citation4].
TFEB (transcription factor EB) is a master regulator of lysosome biogenesis and autophagy [Citation5–7]. Expression, localization, and activity of TFEB is tightly regulated by post-translational modifications, including phosphorylation, ubiquitination, and acetylation [Citation8–10]. Under physiological conditions, TFEB is phosphorylated by MTOR (mechanistic target of rapamycin kinase) complex 1 (MTORC1) on lysosomes and maintained in the cytosol. Upon starvation and cellular stress, MTORC1 is inactivated and/or PPP3/calcineurin phosphatase is activated, leading to the subsequent dephosphorylation and nuclear translocation of TFEB [Citation5]. Nuclear TFEB then binds to a network of promoters (CLEAR; Coordinated Lysosomal Enhancement and Regulation) to induce the transcriptional activation of autophagy and lysosome target genes.
Despite the demonstrated importance of autophagy in EV infections, the exact mechanism by which EVs subvert this pathway, particularly the interplay between EVs and the TFEB network, remains largely unclear. In the current study, we identified TFEB as a novel target of CVB3 that undergoes nuclear translocation, but transcriptional inactivation following infection. Further investigation revealed that TFEB is a bona fide substrate of viral proteinase 3 C, leading to disrupted lysosomal function and enhanced viral propagation.
Results
CVB3 induces nuclear localization but functional attenuation of TFEB
To evaluate the role of CVB3 infection in lysosomal biology, we first investigated the cellular protein TFEB, a master regulator of lysosomal biogenesis and autophagy. TFEB is basally situated in the cytoplasm but undergoes nuclear localization upon activation. HeLa cells were infected with CVB3 (multiplicity of infection [MOI] = 10) following a time-course infection (i.e., 1, 3, or 5 h) to monitor the cellular distribution of TFEB. TFEB was predominantly localized in the cytoplasm in mock-infected cells, while infection with CVB3 caused the re-distribution of TFEB to the nucleus, most evidently at 5 h post-infection (& (b)). It was previously established that nuclear localization of TFEB promotes transcriptional activation of the CLEAR network [Citation11]. To investigate whether CVB3-induced nuclear localization of TFEB similarly activated the CLEAR network, we performed RT-qPCR to analyze several downstream gene targets of TFEB, including M6PR (mannose-6-phosphate receptor, cation dependent), CTSB (cathepsin B) encoding a lysosomal cysteine protease, ATP6V1H (ATPase H+ transporting V1 subunit H) encoding a vacuolar (V)-type proton ATPase subunit, RAB7A (RAB7A, member RAS oncogene family) encoding an endolysosomal trafficking small GTPase, and MCOLN1 (mucolipin 1) encoding a lysosome-associated ion channel. Gene expression for all the aforementioned members of the CLEAR network was significantly reduced, while the mRNA level of house-keeping genes, ACTB and GAPDH, remained unaltered following CVB3 infection ()), indicating impaired expression of TFEB target genes.
Figure 1. CVB3 induces nuclear localization but functional attenuation of TFEB. (a) Intracellular distribution of TFEB in HeLa cells following CVB3 infection. Immunocytochemical staining was performed to examine the localization of TFEB following time-course infection of CVB3 (MOI = 10). Cell nuclei were counterstained with DAPI. Scale bars: 10 µm. (b) Quantification of percentage of TFEB nuclear localization in (A). More than 30 cells were analyzed. (c) mRNA levels of TFEB transcriptional targets in HeLa cells after CVB3 infection. Following 7-h CVB3 infection (MOI = 10), the expression of several TFEB target genes as indicated was measured by real-time quantitative PCR and normalized to ACTB (mean ± SD, n = 3). Transcriptional levels of house-keeping genes ACTB and GAPDH are shown on the right. (d) TFEB transcriptional activity in HeLa cells after CVB3 infection. TFEB transcriptional activity was measured using 4× CLEAR luciferase reporter (mean ± SD, n = 3) following 5-h CVB3 infection (MOI = 10). (e) Protein expression of TFEB in HeLa cells after CVB3 infection. Western blot analysis was conducted to examine protein levels of TFEB following CVB3 infection (MOI = 10) at different time-points. Viral capsid protein (VP1) and ACTB were probed as controls for viral infection and protein loading, respectively. Arrow denotes lower molecular weight protein reactive to TFEB antibody
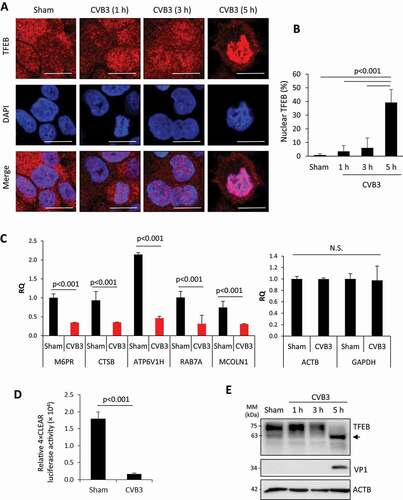
We next examined the effect of CVB3 infection on the transcriptional activity of TFEB, which can be measured using a luciferase reporter construct with 4 tandem CLEAR elements [Citation12]. Consistent with transcriptional inactivation shown in ), the 4× CLEAR luciferase activity was significantly disrupted in CVB3-infected cells ()). We then biochemically assessed TFEB protein in CVB3-infected HeLa lysates. Western blot analysis following various time-points of CVB3 infection revealed a reduction in full-length TFEB and the emergence of a 63-kDa anti-TFEB reactive fragment at 5 h post-infection ()), concomitant with the nuclear translocation of TFEB observed in ) and appearance of the proteolytically processed viral capsid protein VP1 ()). Together, our data suggest that CVB3 infection causes nuclear translocation, but transcriptional inactivation and downregulation of full-length TFEB.
Viral proteinase 3 C cleaves TFEB after Q60
Given that viral polyprotein processing is primarily mediated by the viral proteinase 3 C [Citation13], we next investigated whether the lower-molecular-weight band observed with anti-TFEB antibody was a by-product of 3 C. To that end, we conducted in vitro cleavage assays using purified recombinant wild-type (WT) 3 C or the catalytically inactive (C147A) mutant of 3 C (3 CC147A). ) showed the time-dependent emergence of a 63-kDa anti-TFEB-reactive fragment in the WT 3 C-treated but not catalytically inactive 3 CC147A-incubated condition. The observed fragment in the in vitro reaction was similar in size to that observed following CVB3 time-course infection. Consistent with this, HeLa cells co-transfected with TFEB-GFP and WT 3 C, but not 3 CC147A, recapitulated the appearance of the lower-molecular-weight TFEB fragment ()), further supporting the requirement for 3 C proteinase activity in TFEB cleavage.
Figure 2. Viral proteinase 3 C cleaves TFEB after Q60. (a) Viral proteinase 3 C-mediated cleavage of TFEB by in vitro cleavage assay. HeLa lysates (30 μg) were subjected to in vitro cleavage assay by incubation with 0.1 µg of purified CVB3 wild-type proteinase 3 C (WT 3 C) or catalytically inactive (C147A) mutant of 3 C (3 CC147A) for the indicated times. Cleavage product of TFEB was analyzed by western blotting with anti-TFEB antibody. Arrow denotes cleavage fragment. (b) Viral proteinase 3 C-dependent cleavage of TFEB by ex vivo (cellular) cleavage assay. HeLa cells were transfected with TFEB-GFP together with either empty vector, WT 3 C, or 3 CC147A. After 24 h, cell lysates were collected and analyzed by western blotting with anti-GFP antibody. Arrow denotes TFEB cleavage fragment. (c) Schematic of 3 Cpro consensus cleavage sequence. Q, glutamine; G, glycine; N, asparagine; S, serine; A, alanine. (d) Potential cleavage sites within open reading frame of TFEB. P4-P1ʹ residues are provided in central column. (e-g) Identification of the cleavage site on TFEB. HeLa cells were transfected with WT 3 C or 3 CC147A, together with either TFEBQA15LP/QA28LP (E), TFEBQ112L (f), or TFEBQS60LP, TFEBQS73LP, or TFEBQS86LP (g). After 24 h, cell lysates were harvested and subjected to western blot analysis using anti-TFEB and/or anti-GFP antibodies as indicated. Arrows denote TFEB cleavage fragments. (h) Schematic illustration of the structural domains of TFEB, the identified cleavage site, the antibody recognition regions, and the resulting cleavage products of TFEB
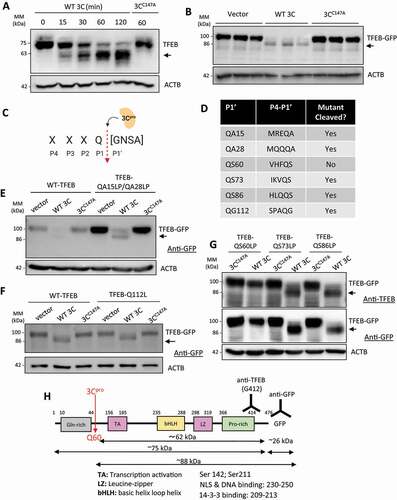
To identify the precise location of proteolytic processing, we screened the TFEB open reading frame for potential cleavage sites with a focus mainly on the highly conserved glutamine (Q) residue at the P1 position ()). We identified 6 potential sites that matched the consensus cleavage residues at the P1-P1ʹ interface with variability being expanded at the P1ʹ position to account for previously reported residues including glycine (G), asparagine (N), serine (S), and alanine (A) [Citation14–16]. Site-directed mutagenesis was used to generate TFEB mutants in the N-terminal region by mutating glutamine (Q) and alanine (A) residues, designated as QA, to leucine (L) and proline (P) residues, which are designated as LP. The resulting mutants included QA15LP QA28LP, Q112L, QS60LP, QS73LP, and QS86LP mutants ()). Co-transfection of WT 3 C with TFEBQA15LP,QA28LP ()), TFEBQ112L ()), TFEBQS73LP or TFEBQS86LP mutants ()) resulted in cleavage of mutant TFEB, suggesting that these sites are not the targets of proteinase 3 C. In contrast, co-expression of WT 3 C with TFEBQS60LP led to complete blockage of 3 C-mediated cleavage ()). Collectively, these data demonstrate that TFEB is cleaved by viral proteinase 3 C after Q60, leading to the generation of a TFEB fragment lacking the N-terminal 60 amino acids, designated as the TFEB [Δ60] fragment ()).
TFEB [Δ60] is non-phosphorylated and nuclear localized
To further study the function of the cleaved TFEB, we generated a GFP-labeled TFEB [Δ60]. Consistent with the results shown in ), WT TFEB was predominantly distributed in the cytoplasm in mock-infected cells and underwent nuclear redistribution upon CVB3 infection. In contrast, TFEB [Δ60] was localized to the nucleus in either mock- or CVB3-infected cells (& (b)).
Figure 3. TFEB [Δ60] is non-phosphorylated and nuclear-localized. (a) Intracellular localization of TFEB and TFEB [Δ60] following CVB3 infection. HeLa cells were transfected with TFEB-GFP or TFEB [Δ60]-GFP for 24 h, followed by CVB3 infection (MOI = 10) for an additional 5 h. GFP fluorescence was used to monitor TFEB localization. Scale bars: 10 µm. (b) Quantification of percentage of TFEB-GFP and TFEB [Δ60]-GFP nuclear localization in (A). More than 30 cells were counted for statistical analysis. (c) Lysosomal localization of TFEB or TFEB [Δ60]. HeLa cells transfected as above were incubated in medium containing the lysosome tracking dye LysoTracker Red DND-99 (75 nM). Scale bars: 10 µm. (d) Quantification of lysosome-localized TFEB-GFP or TFEB [Δ60]-GFP in (C) by ImageJ and presented as Pearson’s correlation coefficient (Rr). More than 30 cells were counted. (e) Dephosphorylation of TFEB following CVB3 infection. HEK293 cells were transfected with either vector, WT-TFEB-GFP, or TFEB [Δ60]-GFP for 24 h, followed by CVB3 infection (MOI = 100) for 8 h. Cell lysates were analyzed by western blotting with anti p-TFEB (Ser211) and anti-ACTB antibodies. (f) Cleavage of exogenous WT-TFEB-GFP following CVB3 infection. HeLa cells were transfected with either WT-TFEB-GFP, TFEBQS60LP-GFP, or TFEB [Δ60]-GFP for 24 h followed by CVB3 infection (MOI = 10) for 8 h. Cell lysates were analyzed by western blotting with anti-GFP antibody. Arrow denotes TFEB cleavage fragment
![Figure 3. TFEB [Δ60] is non-phosphorylated and nuclear-localized. (a) Intracellular localization of TFEB and TFEB [Δ60] following CVB3 infection. HeLa cells were transfected with TFEB-GFP or TFEB [Δ60]-GFP for 24 h, followed by CVB3 infection (MOI = 10) for an additional 5 h. GFP fluorescence was used to monitor TFEB localization. Scale bars: 10 µm. (b) Quantification of percentage of TFEB-GFP and TFEB [Δ60]-GFP nuclear localization in (A). More than 30 cells were counted for statistical analysis. (c) Lysosomal localization of TFEB or TFEB [Δ60]. HeLa cells transfected as above were incubated in medium containing the lysosome tracking dye LysoTracker Red DND-99 (75 nM). Scale bars: 10 µm. (d) Quantification of lysosome-localized TFEB-GFP or TFEB [Δ60]-GFP in (C) by ImageJ and presented as Pearson’s correlation coefficient (Rr). More than 30 cells were counted. (e) Dephosphorylation of TFEB following CVB3 infection. HEK293 cells were transfected with either vector, WT-TFEB-GFP, or TFEB [Δ60]-GFP for 24 h, followed by CVB3 infection (MOI = 100) for 8 h. Cell lysates were analyzed by western blotting with anti p-TFEB (Ser211) and anti-ACTB antibodies. (f) Cleavage of exogenous WT-TFEB-GFP following CVB3 infection. HeLa cells were transfected with either WT-TFEB-GFP, TFEBQS60LP-GFP, or TFEB [Δ60]-GFP for 24 h followed by CVB3 infection (MOI = 10) for 8 h. Cell lysates were analyzed by western blotting with anti-GFP antibody. Arrow denotes TFEB cleavage fragment](/cms/asset/5879cc06-b171-47c1-992d-10b298dc7ae8/kaup_a_1896925_f0003_oc.jpg)
It was previously reported that a subset of cytoplasmic TFEB is anchored to lysosomal membranes and colocalized with LysoTracker Red (LTR, a red-fluorescent dye used as a marker of lysosome or acidic compartments) [Citation17]. Unlike WT-TFEB, we found that TFEB [Δ60] failed to effectively co-localize with lysosomes (& (d)).
The lysosome-associated MTORC1 can suppress TFEB activity through inhibitory phosphorylation on Ser211 [Citation8]. As expected, expression of WT-TFEB resulted in the detection of Ser211 phosphorylation in mock-infected cells, while CVB3 infection led to decreased phosphorylation of WT-TFEB on Ser211 ()). Remarkably, TFEB [Δ60] did not undergo phosphorylation at Ser211 in both mock- and CVB3-infected cells ()). As a control, we showed that the exogenous WT-TFEB was cleaved whereas non-cleavable TFEBQS60LP and the truncated TFEB [Δ60] remained unaffected following CVB3 infection ()). Together, our results reveal that the cleavage fragment of TFEB, TFEB [Δ60], is non-phosphorylated and localized to the nucleus.
TFEB [Δ60] impairs the signaling of lysosomal biogenesis
The nuclear localization nature of TFEB [Δ60] prompted us to evaluate whether this fragment retains the transcriptional activity of WT-TFEB. Following mock infection, cells expressing TFEB [Δ60] had significantly reduced 4× CLEAR transcriptional activity compared to WT-TFEB ()). CVB3 infection further attenuated the transcriptional activity of both WT- and TFEB [Δ60] ()). Most interestingly, we found a significant downregulation in gene targets of TFEB following the expression of TFEB [Δ60] as compared to the expression of vector control in HeLa cells ()), suggesting a possible dominant-negative function of TFEB [Δ60] by competing with the endogenous WT-TFEB.
Figure 4. TFEB [Δ60] impairs the signaling of lysosomal biogenesis. (a) Transcriptional activity of WT-TFEB and TFEB [Δ60]. TFEB transcriptional activity was measured using 4× CLEAR luciferase reporter (mean ± SD, n = 3) following 5 h CVB3 infection (MOI = 10). (b) mRNA levels of TFEB transcriptional targets following expression of TFEB [Δ60]. HeLa cells were transfected with control vector or TFEB [Δ60] for 24 h. Cellular RNA was analyzed by real-time quantitative PCR for the expression of several TFEB target genes as indicated and normalized to ACTB (mean ± SD, n = 3). (c-d) LysoTracker Red (LTR) signals following CVB3 infection (c) or expression of TFEB [Δ60]. HeLa cells were either sham or CVB3 infected (MOI = 10) for 5 h (C) or transfected with control vector or TFEB [Δ60]-GFP for 24 h (d). Nuclear localized TFEB [Δ60] was visualized with GFP fluorescence (D). LTR dye was used as proxy for lysosomal pH measurement and mean LTR intensity was quantified in right panels. More than 30 cells were counted for statistical analysis. Scale bars: 20 μm. (e) LAMP1 signals following expression of WT-TFEB or TFEB [Δ60]. HeLa cells were transiently transfected with WT-TFEB-GFP or TFEB [Δ60]-GFP for 48 h. Cells were fixed and immunostained with anti-LAMP1 antibody. The number of LAMP1-positive structures was visualized by confocal microscopy and quantified from n = 30 cells per condition and presented in the right panel. Scale bars: 20 μm
![Figure 4. TFEB [Δ60] impairs the signaling of lysosomal biogenesis. (a) Transcriptional activity of WT-TFEB and TFEB [Δ60]. TFEB transcriptional activity was measured using 4× CLEAR luciferase reporter (mean ± SD, n = 3) following 5 h CVB3 infection (MOI = 10). (b) mRNA levels of TFEB transcriptional targets following expression of TFEB [Δ60]. HeLa cells were transfected with control vector or TFEB [Δ60] for 24 h. Cellular RNA was analyzed by real-time quantitative PCR for the expression of several TFEB target genes as indicated and normalized to ACTB (mean ± SD, n = 3). (c-d) LysoTracker Red (LTR) signals following CVB3 infection (c) or expression of TFEB [Δ60]. HeLa cells were either sham or CVB3 infected (MOI = 10) for 5 h (C) or transfected with control vector or TFEB [Δ60]-GFP for 24 h (d). Nuclear localized TFEB [Δ60] was visualized with GFP fluorescence (D). LTR dye was used as proxy for lysosomal pH measurement and mean LTR intensity was quantified in right panels. More than 30 cells were counted for statistical analysis. Scale bars: 20 μm. (e) LAMP1 signals following expression of WT-TFEB or TFEB [Δ60]. HeLa cells were transiently transfected with WT-TFEB-GFP or TFEB [Δ60]-GFP for 48 h. Cells were fixed and immunostained with anti-LAMP1 antibody. The number of LAMP1-positive structures was visualized by confocal microscopy and quantified from n = 30 cells per condition and presented in the right panel. Scale bars: 20 μm](/cms/asset/429e6ffe-4419-408c-87ea-cec747c8e7bb/kaup_a_1896925_f0004_oc.jpg)
LTR serves as a functional proxy to assess the luminal acidity required for lysosomal protease activity [Citation18]. Compared to control, we showed that CVB3-infected cells had significantly reduced LTR intensity ()). Furthermore, expression of TFEB [Δ60] alone in the absence of viral infection was able to recapitulate the impairment in lysosomal pH observed following CVB3-infection ()), indicating disruption of the lysosomal function. Finally, we compared the impact of the TFEB [Δ60] mutant on lysosomes using lysosome-associated membrane protein 1 (LAMP1) as a marker. ) showed that compared to WT-TFEB, expression of TFEB [Δ60] mutant is associated with a significant reduction in the number of LAMP1-positive structures.
TFEB [Δ60] retains the ability to interact with CLEAR motif and MITF-family proteins
We next sought to determine the mechanism of TFEB [Δ60] functional deficiency. The transcriptional activity of WT-TFEB relies on its ability to recognize and bind the conserved CLEAR-box sequence (5ʹ-GTCACGTGAC-3ʹ) that is present in the regulatory region of lysosomal target genes ()). Electrophoretic mobility shift assay (EMSA) revealed that similar to WT-TFEB, the TFEB [Δ60] fragment retained its ability to bind the CLEAR E-box sequence (& (c)), consistent with a previous report in which deletion of 120 amino acids of the N-terminus did not impair DNA binding [Citation19]. As expected, co-incubation with anti-TFEB antibody showed reduced mobility shift of the CLEAR probe (& (c)). In addition to nucleotide binding, TFEB transcriptional activity is dependent on hetero- or homodimerization of TFEB with members of the MITF-TFE family of transcription factors [Citation20]. Co-immunoprecipitation studies demonstrated that TFEB [Δ60] could interact with members of the MITF/TFE family, including WT-TFEB ()), MITF-A ()), and TFE3 ()). Collectively, these studies suggest that the functional deficiency of TFEB [Δ60] fragment is not due to impaired CLEAR E-box recognition/binding or hetero/homodimerization with MITF/TFE transcription factors.
Figure 5. TFEB [Δ60] retains the ability to interact with CLEAR and MITF-family proteins. (a) Schematic depiction of WT-TFEB and TFEB [Δ60] in vitro translation and EMSA assay. (b) Binding of WT-TFEB and TFEB [Δ60] to TFEB consensus element. WT-TFEB and TFEB [Δ60] were PCR amplified with T7 promoter for in vitro transcription/translation (left). EMSA assay was performed with TFEB consensus binding element (CLEAR) in the presence of either WT-TFEB, TFEB [Δ60], or WT-TFEB supplemented with anti-TFEB antibody (1 µg). (c) Quantification of EMSA assay in (B). (d-f) Interaction of TFEB [Δ60] with WT-TFEB, MITF-A, and TFE3. HeLa cells were co-transfected with Flag-TFEB [Δ60] and either WT-TFEB-GFP (d), MITF-GFP (e) or TFE3-GFP (f) for 24 h. Immunoprecipitation was conducted with anti-Flag antibody coupled agarose beads, followed by western blot analysis with anti-GFP and anti-Flag antibodies. Densitometry was conducted by ImageJ to quantify the amount of WT-TFEB, MITF-A, and TFE3, normalized to level of antibody IgG light-chain. The fold changes are presented underneath the blots. Western blots of inputs for immunoprecipitation are shown in the right panels
![Figure 5. TFEB [Δ60] retains the ability to interact with CLEAR and MITF-family proteins. (a) Schematic depiction of WT-TFEB and TFEB [Δ60] in vitro translation and EMSA assay. (b) Binding of WT-TFEB and TFEB [Δ60] to TFEB consensus element. WT-TFEB and TFEB [Δ60] were PCR amplified with T7 promoter for in vitro transcription/translation (left). EMSA assay was performed with TFEB consensus binding element (CLEAR) in the presence of either WT-TFEB, TFEB [Δ60], or WT-TFEB supplemented with anti-TFEB antibody (1 µg). (c) Quantification of EMSA assay in (B). (d-f) Interaction of TFEB [Δ60] with WT-TFEB, MITF-A, and TFE3. HeLa cells were co-transfected with Flag-TFEB [Δ60] and either WT-TFEB-GFP (d), MITF-GFP (e) or TFE3-GFP (f) for 24 h. Immunoprecipitation was conducted with anti-Flag antibody coupled agarose beads, followed by western blot analysis with anti-GFP and anti-Flag antibodies. Densitometry was conducted by ImageJ to quantify the amount of WT-TFEB, MITF-A, and TFE3, normalized to level of antibody IgG light-chain. The fold changes are presented underneath the blots. Western blots of inputs for immunoprecipitation are shown in the right panels](/cms/asset/efd5876b-b107-4fe2-85aa-e69ca6d01cc1/kaup_a_1896925_f0005_oc.jpg)
Non-cleavable TFEB rescues 3 C-mediated disruption of TFEB
To test whether non-cleavable TFEB (QS60LP mutant) can rescue the functional deficiency of 3 C-induced cleavage of TFEB, we first examined the cellular localization of TFEBQS60LP following CVB3 infection. Similar to WT-TFEB, non-cleavable TFEB was predominately cytoplasmic during mock infection. However, despite its resistance to cleavage, TFEBQS60LP was significantly trafficked to the nucleus following CVB3 infection ()). Moreover, CVB3 infection resulted in the loss of the lysosomal localization of TFEBQS60LP ()), suggesting that cleavage by viral proteinase 3 C is unlikely the sole factor in regulating TFEB cellular distribution. To evaluate whether non-cleavable TFEB can rescue 3 C-induced functional deficiency of TFEB, we expressed either WT- or TFEBQS60LP in the presence of the viral proteinase 3 C in HeLa cells and found that non-cleavable TFEB was largely resistant to viral proteinase 3 C-mediated disruption of TFEB transcriptional activity ()).
Figure 6. Non-cleavable TFEB rescues 3 C-mediated disruption of TFEB. (a) Nuclear localization of non-cleavable TFEB. HeLa cells were transfected with TFEBQS60LP-GFP for 24 h, followed by sham or CVB3 infection (MOI = 10) for 5 h. Cells were fixed and nuclei were counterstained with DAPI. Percentage of nuclear localization of non-cleavable TFEB was quantified (right). More than 30 cells were selected for the analysis. Scale bar: 20 µm. (b) Lysosomal localization of non-cleavable TFEB. HeLa cells were transfected with TFEBQS60LP-GFP for 24 h, followed by sham or CVB3 infection (MOI = 10) for 5 h in medium containing lysosomal tracking dye LTR (75 nM). Lysosome-localized non-cleavable TFEB was quantified by ImageJ and presented as Pearson’s correlation coefficient (right). More than 30 cells were analyzed. Scale bar: 10 µm. (c) Transcriptional activity of WT- and non-cleavable TFEB following WT 3 C expression. HeLa cells were co-transfected with WT-TFEB or TFEBQS60LP and control vector or WT 3 C with the addition of 4× CLEAR luciferase reporter construct. Luciferase activity was measured (mean ± SD, n = 3). (d) Knockdown of calcineurin/PPP3C by siRNA in HeLa cells. Cells were transfected with either control (si-CON) or PPP3C siRNA (si-PPP3C) for 48 h. Knockdown efficiency was validated by western blot analysis with anti-PPP3C antibody. (e) Impairment of CVB3-induced trafficking of TFEB after gene-silencing of PPP3C. HeLa cells transfected with si-CON or si-PPP3C as above were infected with CVB3 (MOI = 10) for 5 h. GFP-labeled TFEBQS60LP was analyzed by immunofluorescence. Nuclei were counterstained with DAPI. Nuclear TFEB was quantified in the right panel (number of analyzed cells >30). Scale bars: 10 μm. (f) Rescue of CVB3-induced dephosphorylation of TFEBQS60LP after gene-silencing of PPP3C. HeLa cells expressing TFEBQS60LP were treated with either si-CON or si-PPP3C for 48 h prior to CVB3 infection (MOI = 10) for 5 h. Cell lysates were subjected to western blot analysis of p-TFEB (Ser211), TFEBQS60LP-GFP, VP1 and ACTB. Densitometry was conducted to analyze the ratio of p-TFEB to TFEB and VP1:ACTB. Results (mean ± SD, n = 3) are presented in the right panel
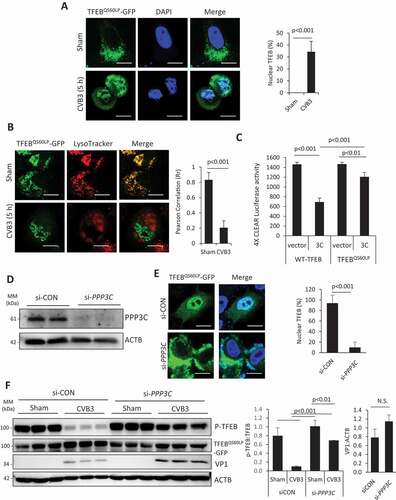
The re-distribution of TFEBQS60LP to the nucleus following CVB3 infection prompted us to evaluate the mechanism of cleavage-independent trafficking of TFEB. Dephosphorylation of TFEB by the calcium responsive phosphatase calcineurin was previously shown to regulate TFEB nuclear trafficking following lysosomal damage [Citation5]. To investigate the potential involvement of calcineurin in CVB3-induced TFEB dephosphorylation, we infected TFEBQS60LP expressing cells treated with either siRNA targeting PPP3C (the catalytic subunit of calcineurin) or scrambled siRNA with CVB3. Protein expression of PPP3C was confirmed to be markedly reduced in siRNA-treated cells compared to scrambled control ()). In a parallel experiment, the distribution of non-cleavable TFEBQS60LP was monitored following CVB3 infection. Compared to control cells, calcineurin-depleted cells had significantly reduced nuclear TFEBQS60LP ()) and elevated phospho(p)-Ser211 ()) without apparent alteration in viral replication as measured by VP1 expression ()). Collectively, these results suggest that, in addition to processing TFEB, CVB3 infection also triggers a calcineurin-dependent trafficking of WT-TFEB to the nucleus.
TFEB [Δ60] enhances viral infection
EVs, such as CVB3, rely on various cellular factors for effective replication. To investigate the role of TFEB in viral infection, HeLa cells were transfected with siRNA targeting TFEB (si-TFEB) or scrambled siRNA (si-CON) for 48 h. Cells were subsequently infected with CVB3 (MOI = 10) for an additional 7 h and the supernatant was collected for measurement of viral titers. Compared to control treatment, gene-silence of TFEB resulted in a significant attenuation of extracellular CVB3 titers without evident impacts on viral protein production ()). To exclude the potential involvement of TFEB in viral entry, cells treated with si-CON or si-TFEB were incubated with CVB3 for 1.5 h. After thorough washes, cellular RNA was harvested and assessed for viral RNA expression. It was observed that two groups of cells had comparable post-entry viral RNA, suggesting that TFEB is dispensable for CVB3 entry ()).
Figure 7. TFEB [Δ60] enhances viral infection. (a) Reduced viral titers after gene-silence of TFEB. HeLa cells were treated with si-CON or si-TFEB for 48 h, followed by CVB3 infection (MOI = 10) for 7 h. Extracellular medium was harvested and viral titers were analyzed by TCID50 (mean ± SD, n = 3. left). Cell lysates were subjected to western blotting with anti-TFEB and anti-VP1 antibodies (middle). Densitometry was performed and the fold changes of TFEB and VP1 over ACTB are presented underneath the blots. VP1:ACTB is also shown in the bar plot (mean ± SD, n = 3, right). Arrow denotes TFEB cleavage fragment. (b) No impact on CVB3 entry after gene-silence of TFEB. HeLa cells were treated with si-CON or si-TFEB as in (A). Cells were incubated with CVB3 for 1.5 h, rinsed thrice with PBS, and subjected to RNA extraction. Viral RNA was assessed by RT-qPCR (mean ± SD, n = 3). (c) Enhanced CVB3 titers after overexpression of TFEB [Δ60]. HeLa cells were transfected with control vector, WT-TFEB, TFEB [Δ60], or TFEBQS60LP for 24 h. Cells were subsequently infected with CVB3 (MOI = 0.1) for 24 h. Viral titers in the supernatant were analyzed by TCID50 (mean ± SD, n = 3). Protein expression was verified by western blotting using anti-GFP antibody. (d) Schematic illustration of membrane integrity probe following CVB3 infection. (e) HeLa cells were sham-infected or infected with CVB3 for the indicated time-points and stained with the LIVE/DEAD membrane integrity probe. Cells were fixed and visualized under confocal microscopy with an excitation wavelength of 580 nm. Scale bar: 20 µm. (f) Enhanced non-lytic release after overexpression of TFEB [Δ60]. HeLa cells were transfected with si-CON or si-TFEB for 48 h, followed by reconstitution with either control vector, WT-TFEB, or TFEB [Δ60], for 24 h. Cells were subsequently infected with CVB3 for 5 h. Viral titers in the supernatant were analyzed by TCID50 (mean ± SD, n = 3) as a measure of non-lytic egress
![Figure 7. TFEB [Δ60] enhances viral infection. (a) Reduced viral titers after gene-silence of TFEB. HeLa cells were treated with si-CON or si-TFEB for 48 h, followed by CVB3 infection (MOI = 10) for 7 h. Extracellular medium was harvested and viral titers were analyzed by TCID50 (mean ± SD, n = 3. left). Cell lysates were subjected to western blotting with anti-TFEB and anti-VP1 antibodies (middle). Densitometry was performed and the fold changes of TFEB and VP1 over ACTB are presented underneath the blots. VP1:ACTB is also shown in the bar plot (mean ± SD, n = 3, right). Arrow denotes TFEB cleavage fragment. (b) No impact on CVB3 entry after gene-silence of TFEB. HeLa cells were treated with si-CON or si-TFEB as in (A). Cells were incubated with CVB3 for 1.5 h, rinsed thrice with PBS, and subjected to RNA extraction. Viral RNA was assessed by RT-qPCR (mean ± SD, n = 3). (c) Enhanced CVB3 titers after overexpression of TFEB [Δ60]. HeLa cells were transfected with control vector, WT-TFEB, TFEB [Δ60], or TFEBQS60LP for 24 h. Cells were subsequently infected with CVB3 (MOI = 0.1) for 24 h. Viral titers in the supernatant were analyzed by TCID50 (mean ± SD, n = 3). Protein expression was verified by western blotting using anti-GFP antibody. (d) Schematic illustration of membrane integrity probe following CVB3 infection. (e) HeLa cells were sham-infected or infected with CVB3 for the indicated time-points and stained with the LIVE/DEAD membrane integrity probe. Cells were fixed and visualized under confocal microscopy with an excitation wavelength of 580 nm. Scale bar: 20 µm. (f) Enhanced non-lytic release after overexpression of TFEB [Δ60]. HeLa cells were transfected with si-CON or si-TFEB for 48 h, followed by reconstitution with either control vector, WT-TFEB, or TFEB [Δ60], for 24 h. Cells were subsequently infected with CVB3 for 5 h. Viral titers in the supernatant were analyzed by TCID50 (mean ± SD, n = 3) as a measure of non-lytic egress](/cms/asset/3b57af2e-a275-4d84-846c-15162db19bcf/kaup_a_1896925_f0007_oc.jpg)
To further evaluate the functional consequence of TFEB cleavage in viral infection, HeLa cells were transiently transfected with constructs expressing either WT-TFEB, TFEB [Δ60], TFEBQS60LP, or vector control for 24 h, followed by CVB3 infection (MOI = 0.1) for an additional 24 h. A lower MOI and longer duration of infection was used to discern any subtle changes in viral propagation. We demonstrated that expression of TFEB [Δ60] was associated with a significant enhancement of viral titers, which appeared to be more evident than WT- and non-cleavable TFEB at this given MOI and length of viral infection ()), suggesting a pro-viral function for the cleaved TFEB fragment. To assess the role of TFEB in non-lytic egress, we monitored cell membrane integrity following CVB3 infection with the amine-reactive LIVE/DEAD dye. These dyes can penetrate dead cells and fluoresce via laser excitation following chemical reactions with free amines ()). During sham infection, the dye fluoresced on the surface of cells whereas prolonged CVB3 infection at 7 h and 9 h resulted in strong cytoplasmic signal, suggesting disrupted membrane integrity at these time-points ()). Since the membrane integrity of cells was comparable between sham and 5 h infection, 5 h post-infection was selected for measurement of non-lytic egress. HeLa cells pre-treated with either control or si-TFEB for 48 h were subsequently reconstitution with WT-TFEB or TFEB [Δ60]. Following 24 h, cells were infected with CVB3 (MOI = 10) for an additional 5 h and supernatants were collected for viral titer measurements. We showed that gene-silence of TFEB resulted in attenuated egress of CVB3 compared to control cells whereas expression of WT-TFEB was able to rescue this deficit ()). Of note, expression of TFEB [Δ60] was associated with enhanced egress of CVB3. Altogether, our data suggest that TFEB and TFEB [Δ60] have roles in viral egress albeit through differing mechanisms.
Discussion
The subversion of cellular autophagy by EVs has become a topic of great interest ever since the first reported studies documenting the proliferation of double-membraned vesicles reminiscent of autophagosomes following EV infection [Citation21,Citation22]. The normal function of autophagosomes is to enwrap cellular cargo, such as protein aggregates, damaged organelles, and invading pathogens, for clearance in lysosomes; but certain pathogens, such as EVs, have evolved strategies to evade the degradative capacity of autophagy. For example, CVB3 and EV-D68 can utilize viral proteinase 3 C to target autophagic SNAREs that are required for autophagosomes-lysosome fusion for viral benefits [Citation23,Citation24]. The current study identifies an additional mechanism by which EVs evade lysosomal degradation. Lysosomes are increasingly being recognized as more than a disposal chamber for cellular waste, being involved in various cellular processes from signaling and energy metabolism to secretions and membrane repair [Citation25,Citation26]. We demonstrate that the viral proteinase 3 C of CVB3 targets the master transcriptional regulator of lysosome biogenesis, TFEB, to impair lysosomal function and enhance viral propagation.
Following CVB3 infection, TFEB is re-distributed to the nucleus but paradoxically, gene expression of downstream targets is significantly attenuated. It was further identified that TFEB is cleaved by viral proteinase 3 C at the N-terminus following Q60 to generate a fragment, designated as TFEB [Δ60]. This fragment retains the capacity to recognize and bind CLEAR-box elements as well as homo- and heterodimerize with members of the basic helix-loop-helix-leucine-zipper (bHLH-Zip) family of MITF/TFE transcription factors, but loses its function in transcriptional activation. Since TFEB [Δ60] lacks the Q-rich domain (residues 10–44), we speculate that functional deficiency in TFEB [Δ60] is related to this missing domain. Indeed, Q-rich domains are highly enriched in eukaryotic transcription factors and have been shown to modulate transcriptional activation [Citation27]. It was reported that addition of homopolymeric stretches of the Q residues activates transcription when fused to the DNA binding domain of GAL4 [Citation28]. The precise cleavage of TFEB to eliminate its transactivation capacity while retaining its binding capabilities may be an unappreciated viral strategy to generate a dominant negative function. Additionally, the attenuation of TFEB transactivation may contribute to the downregulation of downstream CLEAR genes such as the vacuolar H+ ATPase, ATP6V1H, which acts to facilitate lysosome acidification.
Subcellular localization and activity of TFEB is controlled by its phosphorylation status at key residues such as Ser211. p-Ser211 serves as a docking site for the YWHA/14-3-3 family of chaperones that sequester TFEB in the cytosol [Citation8]. Phosphorylation of TFEB is mediated by MTORC1, which takes place at the lysosomal surface [Citation29]. Our study reveals that CVB3 infection promotes the dephosphorylation of TFEB, consistent with recent findings by Alirezaei et al [Citation30]. TFEB harbors a lysosome localization signal (LLS) within the first 30 amino acids at its N-terminus that mediates lysosomal localization of TFEB and consequent phosphorylation [Citation8]. We therefore postulate that cleavage of TFEB after Q60 and the subsequent loss of the LLS contribute to the nuclear re-localization of TFEB observed following CVB3 infection. Interestingly, we found that, in addition to the cleavage fragment, non-cleavable TFEB is also re-localized to the nucleus following CVB3 infection, suggesting that CVB3 triggers additional mechanisms that facilitate nuclear redistribution of TFEB. Lysosomal calcium release was reported to activate the serine-threonine phosphatase calcineurin and promote the subsequent dephosphorylation of its substrate TFEB [Citation5]. Using non-cleavable TFEB (QS60LP), we demonstrate that nuclear re-distribution of TFEB is partially mediated by calcineurin in a cleavage-independent manner. A proposed model of CVB3-induced dysregulation of TFEB and role in viral egress is summarized in .
Figure 8. Proposed model of TFEB dysregulation following CVB3 infection. (1) TFEB is dephosphorylated in a calcineurin-dependent manner following CVB3 infection. (2) TFEB is cleaved by viral proteinase 3 C to generate TFEB [Δ60]. (3) TFEB [Δ60] interacts with TFEB and other members of MITF/TFE family as well as CLEAR elements. (4) TFEB [Δ60] is associated with attenuation of CLEAR network. (5) TFEB and TFEB [Δ60] play a role in non-lytic egress of CVB3
![Figure 8. Proposed model of TFEB dysregulation following CVB3 infection. (1) TFEB is dephosphorylated in a calcineurin-dependent manner following CVB3 infection. (2) TFEB is cleaved by viral proteinase 3 C to generate TFEB [Δ60]. (3) TFEB [Δ60] interacts with TFEB and other members of MITF/TFE family as well as CLEAR elements. (4) TFEB [Δ60] is associated with attenuation of CLEAR network. (5) TFEB and TFEB [Δ60] play a role in non-lytic egress of CVB3](/cms/asset/db5d7d1f-2d0a-41aa-9ffe-932889059129/kaup_a_1896925_f0008_oc.jpg)
EVs have evolved to fine-tune efficient replication by differentially regulating host factors at various stages of the viral replication cycle. Certain host factors may be required during an early replicative stage but become dispensable or anti-viral during late infection [Citation31]. In the present study, we demonstrate that gene-silencing of TFEB prior to infection leads to the attenuation of CVB3 titers, supporting recent evidence that TFEB serves as a pro-viral factor to facilitate viral replication [Citation30] and non-lytic egress [Citation32] at early viral infection. Interestingly, we discovered that during late stage of viral infection, TFEB is proteolytically processed and the cleavage fragment further enhances viral propagation. Our finding suggests that CVB3 has evolved to co-opt TFEB to shut-off its transcriptional activity and lysosomal repair mechanisms as a viral strategy to evade lysosomal degradation during late viral infection. The evidence of impaired transactivation by TFEB following CVB3 infection coupled with cleaved TFEB [Δ60] retaining its capacity to interact with full-length TFEB, likely supports that CVB3 may employ a two-pronged approach to incapacitate TFEB: (1) cleaved TFEB [Δ60] traffics to the nucleus to impair CLEAR network activation and/or (2) TFEB [Δ60] complexes with full-length TFEB to impair its transactivation capacity. Both outcomes result in the downregulation of lysosomal signaling that may ultimately favor CVB3 subversion of autophagy. EV proteinases are reported to target many host substrates leading to complex functional interplay within infected cells [Citation15]. Although the current study characterizes TFEB as a novel substrate of viral proteinase 3 C, the complex interplay this event generates with other cleaved host-proteins still merits further study.
In summary, our study reveals that CVB3 directly targets TFEB for proteolytic cleavage and transcriptional inactivation, leading to impaired lysosomal function and increased viral propagation. These insights ultimately provide a better understanding of how EVs subvert cellular autophagy to promote viral pathogenesis.
Materials and methods
Cell culture and viral infection
HeLa and HEK293 cells (American Type Culture Collection, CCL-2 and CRL-1573) were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and a penicillin-streptomycin cocktail (100 µg/mL). For CVB3 infection, cells were either sham-infected with PBS (Sigma, D8537) or inoculated with CVB3 (Kandolf strain; generously provided by Reinhard Kandolf, University of Tubingen, Germany) at different MOIs as specified in the figure legends.
Plasmids and small interfering RNA (siRNA)
The 4× CLEAR luciferase reporter (66,800), pEGFP-N1-TFEB (38,119), pEGFP-N1-MITF-A (38,132), and pEGFP-N1-TFE3 (38,120) plasmids were obtained from Addgene (deposited by Albert La Spada and Shawn Ferguson). pEGFP-N1-TFEB [Δ60] plasmid was generated in the Luo lab. TFEB [Δ60] insert was amplified by PCR with Forward primer: AAATTTAAGCTTGCCACC ATGGGATCGCCACCACCTGTGC and Reverse primer: AAATTTGGTACCTTCAGCACATCGCCCTCCTC and cloned into pEGFP-N1 at HindIII and KpnI sites. 3× Flag-TFEB [Δ60] was generated in the current laboratory. TFEB [Δ60] insert was amplified by PCR with Forward primer: AAATTTGAATTCCTCGCCACCACCTGTGC and Reverse primer: AAATTTGGATCCTCACAGCACATCGCCCTCCTC and cloned into CMV10 vector at EcoRI and BamHI sites. TFEBQS60LP, TFEBQS73LP, and TFEBQS86LP were generated through site-directed mutagenesis with the following primers (QS60LP Forward: 5ʹCCGTCCACTTCCTGCCGCCACCAC-3ʹ Reverse: 5ʹ GTGGTGGCGGCAGGAAGTGGACGG-3ʹ; QS73LP Forward: 5ʹ GTGTTGAAGGTGCTGCCCTACCTGGAG-3ʹ Reverse: 5ʹ CTCCAGGTAGGGCAGCACCTTCAACAC-3ʹ; QS86LP Forward: 5ʹ CTACCATCTGCAGCTGCCGCAGCATCAG-3ʹ Reverse: 5ʹ CTGATGCTGCGGCAGCTGCAGATGGTAG-3ʹ). The MYC-tagged wild-type CVB3-3 C (WT 3 C) and C147A mutant CVB3-3 C (3 CC147A) constructs were generous gifts from Dr. Carolyn Coyne at the University of Pittsburgh [Citation33].
The siRNA targeting TFEB (sc-38,509) and scrambled siRNA (sc-37,007) were purchased from Santa Cruz Biotechnology. For transfection, cells were transiently transfected with plasmid cDNAs or siRNAs using Lipofectamine 2000 (Invitrogen, 11,668–019) following the manufacturer’s instructions.
Confocal microscopy
HeLa cells were cultured in 8-well chamber slides (Labtek, 155,411) for 24 h prior to treatment.
Cell membrane integrity was assessed with the amine-reactive dye LIVE/DEAD (Thermo Fisher Scientific, L34958) according to the manufacturer’s instructions. After fixation in 4% paraformaldehyde, cells were washed thrice in PBS. For LAMP1 staining, cells were permeabilized with 0.1% Triton X-100 (Sigma, T8787), blocked with 3% BSA (Sigma, A7030), and incubated overnight with Alexa Fluor 594-conjugated anti-LAMP1 (Santa Cruz Biotechnology, sc20011) before mounting with fluoroshield with 4, 6-diamidino-2-phenylindole (DAPI; Sigma-Aldrich, F6057). Images were captured with the Zeiss LSM 880 Inverted Confocal Microscopy using a 63× objective lens.
LysoTracker staining
Lysosomal colocalization was monitored using LysoTracker Red (LTR) DND-99 (Thermo Fisher Scientific, L7528) following the manufacturer’s guideline. Briefly, HeLa cells were cultured in 8-well chamber slides (Labtek, 155,411) for 24 h prior to treatment. Following construct transfection (24 h) and/or CVB3 infection (5 h), LTR (75 nM) was added into the culture medium and incubated at 37°C for 60 min. Images were captured with the Zeiss LSM 880 Inverted Confocal Microscopy using a 63× oil immersion lens. LTR mean intensity was analyzed using ImageJ software.
4× CLEAR luciferase assay
TFEB transcriptional activity was measured using the dual-luciferase reporter system (Promega, E1910). Briefly, HeLa cells were co-transfected with 4× CLEAR luciferase reporter (Addgene, 66,800; deposited by Albert La Spada) and Renilla control luciferase plasmid (Addgene, 87,121; deposited by Sanjiv Sam Gambhir). Cells were harvested at 16 h post transfection, and cell lysates were transferred to an opaque 96-well microplate (Greiner, 655,074). Luciferase activity was detected using SpectraMax i3 Detection System.
In vitro cleavage assay
In vitro cleavage assay was performed as previously described [Citation34]. Briefly, HeLa lysates (20 μg) were incubated with CVB3 WT 3 C or catalytically inactive (C147A) 3 C [mut] (0.1 μg) in cleavage assay buffer (20 mM HEPES, pH 7.4, 150 mM KOAc, 1 mM DTT) for the indicated times at 37°C. Reactions were terminated with 6× sample buffer followed by 95°C denaturation and subsequent western blot analysis.
Western blot analysis
Cells were lysed in buffer (10 mM HEPES, pH 7.4, 50 mM NaPyrophosphate, 50 mM NaF, 50 mM NaCl, 5 mM EDTA, 5 mM EGTA, 100 µM Na3VO4, 0.1% Triton X-100) and western blotting was conducted using the following primary antibodies: TFEB (Cell Signaling Technology, D2O7D), phospho (Ser211)-TFEB (Cell Signaling Technology, 376,815), VP1 (Mediagnost, Cox mAb 31A2), ACTB (Sigma-Aldrich, A5316), GFP (Sigma-Aldrich, SAB5300167), FLAG (Sigma, F1804), and PPP3C/calcineurin (Santa Cruz Biotechnology, sc-17,808).
Ex vivo cleavage assay
Ex vivo or cellular cleavage assay was performed as previously described [Citation34]. Briefly, HeLa cells were transfected with TFEBQS60LP, TFEBQS73LP, or TFEBQS86LP and either CVB3 WT 3 C or catalytically inactive (C147A) mutant 3 Cmut for 16 h. Lysates were harvested and subjected to western blot analysis with anti-GFP and anti-TFEB antibody to detect cleavage fragments.
Electrophoretic mobility shift assay (EMSA)
In vitro transcription/translation of TFEB and TFEB [Δ60] were performed using the TNT® T7 Quick Coupled Transcription/Translation System (Promega, L1170) according to the manufacturer’s protocol. EMSA was carried out in 20 µl reaction containing 1 µl of reticulocyte translation product in binding buffer (5% glycerol, 100 mM KCl, 10 mM MgCl2, 10 mM Tris, pH 7.4, 1 mM DTT) supplemented with 0.5 ng of TFEB consensus oligonucleotide (Santa Cruz Biotechnology, sc-2621). Control reaction was supplemented with 1 μg of anti-TFEB antibody (Cell Signaling Technology, D2O7D). Reaction mixtures were incubated at room temperature for 20 min. and separated by 6% non-denaturing polyacrylamide gel. Nucleic acid was visualized with SYBR™ Safe DNA Gel Stain (Thermofisher Scientific, S33102) and imaged using the GBOX Chemi XRQ.
Immunoprecipitation
Immunoprecipitation (IP) of Flag-tagged TFEB [Δ60] was performed using EZviewTM Red ANTI-FLAG ® M2 Affinity Gel (Sigma-Aldrich, F2426) according to the manufacturer’s instructions. In brief, HeLa cell lysates were incubated with anti-Flag M2 agarose beads at 4°C overnight. After three washes, the bound proteins were eluted with 2× SDS sample buffer and then subjected to western blot analysis.
Viral titer measurement
Samples were serially diluted and overlaid on 60-well Terasaki plates (Sarstedt, 83.9923.972) of HeLa cells. After 48 h incubation, 50% tissue culture infective dose titer (TCID50) was calculated by the statistical method of Reed and Muench [Citation35]. Viral titers are expressed as plaque-forming unit (PFU)/mL with 1 infectious unit equal to 0.7 TCID50 as described previously [Citation36].
Viral entry assay
Viral particle uptake was performed as described previously [Citation37]. In brief, HeLa cells treated with control or TFEB-targeting siRNA were infected with CVB3 for 1.5 h. After three washes with PBS, cells were harvested and subjected to RNA extraction. qPCR was performed to determine levels of viral genomic RNA using primer pairs of CVB3 2A (forward: 5′-GCT TTG CAG ACA TCC GTG ATC-3′; reverse: 5′-CAA GCT GTG TTC CAC ATA GTC CTT CA-3′) and normalized to ACTB/β-actin mRNA (ACTB, forward primer: 5′-ACT GGA ACG GTG AAG GTG AC-3′; reverse primer: 5′-GTG GAC TTG TTG GGA GAG GAC TG-3′) levels.
Real-time quantitative RT-PCR
Total RNA was extracted using the RNeasy Mini kit (Qiagen, 74,104). To determine gene expression levels, quantitative PCR targeting M6PR (forward primer: CTCAGTGTGGGTTCCATCTTAC; reverse primer: GGGAAACTGCTCCATTCCTT), CTSB (forward primer: GGACAAGCACTACGGATACAA; reverse primer: GTAGAGCAGGAAGTCCGAATAC) ATP6V1H (forward primer: CCCTGAAGAGAAGCAAGAGATG, reverse primer: TGCAGCATATCATCCACCATAG)
RAB7A (forward primer: CCTGGAGTCTTGGCCATAAAG, reverse primer: GAGAAGGTCCAAGTTCTGGTTC) MCOLN1 (forward primer: GGAAAGCAGCTCCAGTTACA reverse primer: GATGAGGCTCTGGAGGTTAATG) was performed in a 10 µl reaction containing 1 μg of RNA using the TaqMan™ RNA-to-CT™ 1-Step Kit (Life Technologies, 4,392,653) and normalized to ACTB mRNA according to the manufacturer’s instructions. The PCR reaction was performed on a ViiA 7 Real-Time PCR System (Applied Biosystems). Samples were run in triplicate and analyzed using comparative CT (2-ΔΔCT) method with control samples and presented as relative quantitation (RQ).
Statistical analysis
Results are presented as mean ± standard deviation (SD). Statistical analysis was performed with unpaired Student’s t-test to compare two groups or analysis of variance (ANOVA) with Tukey post-hoc test using GraphPad Prism 5 software to evaluate multiple groups. A P-value <0.05 was considered to be statistically significant. All results presented are representative of at least 3 independent experiments.
Disclosure statement
All authors declare that they have no conflict of interest.
Additional information
Funding
References
- Khetsuriani N, Lamonte-Fowlkes A, Oberst S, et al. Centers for disease C, prevention. Enterovirus surveillance–United States, 1970-2005. MMWR Surveill Summ. 2006;55:1–20.
- Baggen J, Thibaut HJ, Strating JRPM, et al. The life cycle of non-polio enteroviruses and how to target it (vol 16, pg 368, 2018). Nat Rev Microbiol. 2018;16:391.
- Mohamud Y, Luo H. The intertwined life cycles of enterovirus and autophagy. Virulence. 2019;10:470–480.
- Lamming DW, Bar-Peled L. Lysosome: the metabolic signaling hub. Traffic. 2019;20:27–38.
- Medina DL, Di Paola S, Peluso I, et al. Lysosomal calcium signalling regulates autophagy through calcineurin and TFEB. Nat Cell Biol. 2015;17:288–299.
- Napolitano G, Ballabio A. TFEB at a glance. J Cell Sci. 2016;129:2475–2481.
- Settembre C, Di Malta C, Polito VA, et al. TFEB links autophagy to lysosomal biogenesis. Science. 2011;332:1429–1433.
- Roczniak-Ferguson A, Petit CS, Froehlich F, et al. The transcription factor TFEB links mTORC1 signaling to transcriptional control of lysosome homeostasis. Sci Signal. 2012;5:ra42.
- Sha YB, Rao L, Settembre C, et al. STUB1 regulates TFEB-induced autophagy-lysosome pathway. Embo J. 2017;36:2544–2552.
- Zhang JB, Wang JG, Zhou ZH, et al. Importance of TFEB acetylation in control of its transcriptional activity and lysosomal function in response to histone deacetylase inhibitors. Autophagy. 2018;14:1043–1059.
- Settembre C, Zoncu R, Medina DL, et al. A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. Embo J. 2012;31:1095–1108.
- Cortes CJ, Miranda HC, Frankowski H, et al. Polyglutamine-expanded androgen receptor interferes with TFEB to elicit autophagy defects in SBMA. Nat Neurosci. 2014;17:1180–1189.
- Sun D, Chen S, Cheng AC, et al. Roles of the picornaviral 3C proteinase in the viral life cycle and host cells. Viruses-Basel 2016; 8.
- Jagdeo JM, Dufour A, Klein T, et al. N-terminomics TAILS identifies host cell substrates of poliovirus and Coxsackievirus B3 3C proteinases that modulate virus infection. J Virol. 2018;92(8):e02211-17.
- Saeed M, Kapell S, Hertz NT, et al. Defining the proteolytic landscape during enterovirus infection. Plos Pathog. 2020;16(9):e1008927.
- Blom N, Hansen J, Blaas D, et al. Cleavage site analysis in picornaviral polyproteins: discovering cellular targets by neural networks. Protein Sci. 1996;5:2203–2216.
- Bampton ETW, Goemans CG, Niranjan D, et al. The dynamics of autophagy visualized in live cells - From autophagosome formation to fusion with endo/lysosomes. Autophagy. 2005;1:23–36.
- Kliosnky D. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition).Autophagy. 2016;12(1):1–222.
- Fisher DE, Carr CS, Parent LA, et al. Tfeb has DNA-binding and oligomerization properties of a unique helix loop helix leucine-zipper family. Gene Dev. 1991;5:2342–2352.
- Slade L, Pulinilkunnil T. The MiTF/TFE family of transcription factors: master regulators of organelle signaling, metabolism, and stress adaptation. Mol Cancer Res. 2017;15:1637–1643.
- Dales S, Eggers HJ, Tamm I, et al. Electron microscopic study of the formation of poliovirus. Virology. 1965;26:379–389.
- Horne RW, Nagington J. Electron microscope studies of the development and structure of poliomyelitis virus. J Mol Biol. 1959;1:333.
- Corona AK, Saulsbery HM, Velazquez AFC, et al. Enteroviruses remodel autophagic trafficking through regulation of host SNARE proteins to promote virus replication and cell exit. Cell Rep. 2018;22:3304–3314.
- Mohamud Y, Shi J, Qu J, et al. Enteroviral infection inhibits autophagic flux via disruption of the SNARE complex to enhance viral replication. Cell Rep. 2018;22:3292–3303.
- Reddy A, Caler EV, Andrews NW. Plasma membrane repair is mediated by Ca2+-regulated exocytosis of lysosomes. Cell. 2001;106:157–169.
- Settembre C, Fraldi A, Medina DL, et al. Signals from the lysosome: a control centre for cellular clearance and energy metabolism. Nat Rev Mol Cell Biol. 2013;14:283–296.
- Gemayel R, Chavali S, Pougach K, et al. Variable glutamine-rich repeats modulate transcription factor activity. Mol Cell. 2015;59:615–627.
- Gerber HP, Seipel K, Georgiev O, et al. Transcriptional activation modulated by homopolymeric glutamine and proline stretches. Science. 1994;263:808–811.
- Sancak Y, Bar-Peled L, Zoncu R, et al. Ragulator-rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell. 2010;141:290–303.
- Alirezaei M, Flynn CT, Garcia SD, et al. A food-responsive switch modulates TFEB and autophagy, and determines susceptibility to coxsackievirus infection and pancreatitis. Autophagy. 2020. DOI: https://doi.org/10.1080/15548627.2020.1720425
- Lloyd RE. Nuclear proteins hijacked by mammalian cytoplasmic plus strand RNA viruses. Virology. 2015;479:457–474.
- Giansanti P, Strating J, Defourny KAY, et al. Dynamic remodelling of the human host cell proteome and phosphoproteome upon enterovirus infection. Nat Commun. 2020;11:4332.
- Harris KG, Coyne CB. Unc93b induces apoptotic cell death and is cleaved by host and enteroviral proteases. Plos One. 2015;10(10): e0141383.
- Mohamud Y, Qu J, Xue YC, et al. CALCOCO2/NDP52 and SQSTM1/p62 differentially regulate coxsackievirus B3 propagation. Cell Death Differ. 2019;26:1062–1076.
- Reed LJ, Muench H. A simple method of estimating fifty percent endpoints. Am J Hyg. 1938;27:493–497.
- Davis BD, Dulbecco R, Eisen HN, et al. Nature of viruses. In: Microbiology. New York: Harper and Row; 1972. p. 1044–1053.
- Deng HY, Liu HT, De Silva T, et al. Coxsackievirus type B3 is a potent oncolytic virus against KRAS-mutant lung adenocarcinoma. Mol Ther Oncolytics. 2019;14:266–278.