
ABSTRACT
Although it has been reported that some autophagy-related proteins could regulate the cell cycle, the function of ULK1-ATG13, the only protein kinase complex in macroautophagy/autophagy, remains unclear. We recently found that mitotic ULK1 and ATG13 are both substrates of the key cell cycle regulator CDK1-CCNB/cyclin B. CDK1-induced ULK1-ATG13 phosphorylation promotes mitotic autophagy and cell cycle progression. Moreover, ULK1 and ATG13 double-knockout significantly inhibits cell cycle progression and tumor cell proliferation in vitro and in vivo. These findings bridge the mutual regulation between autophagic and mitotic key kinases and provide a theoretical basis for autophagy- and cell division-related diseases based on combination therapy.
Autophagy regulation mechanisms were mainly investigated in asynchronous cells. However, it was recently reported by multiple groups that autophagy is differentially regulated throughout the cell cycle, especially in the short but very dynamic cell cycle phase, mitosis. Although it has been shown that the autophagic flux remains active in mitosis, and multiple kinases such as CDKs (cyclin dependent kinases), AURKs (aurora kinases), PLK1 (polo like kinase 1), MTOR (mechanistic target of rapamycin kinase) complex 1 (MTORC1), and AMPK and ATG proteins such as ATG7, RB1CC1/FIP200, BECN1/Beclin-1, and ATG5 could play dual roles in both autophagy and cell-cycle or mitosis, the exact underlying regulation mechanism in mitosis, or the whole cell cycle, is still underexplored. Moreover, whether ULK1-ATG13, the core machinery for the ULK1 autophagy initiation complex, also participates in cell-cycle and mitosis regulation has not been investigated.
We found an obvious electrophoretic bandshift of endogenous and exogenous ULK1-ATG13 in mitosis, but not ATG5, BECN1, or ATG101 () [Citation1]. Because bandshift implies proline-directed phosphorylation, we used a general anti-phosphoserine/threonine antibody or lambda phosphatase to confirm that ULK1-ATG13 is hyperphosphorylated in mitosis. The kinase inhibitor screen suggested CDK1 as its potential kinase, and immunoprecipitation revealed that CDK1-CCNB1 associates with ULK1-ATG13 in vivo. Importantly, ULK1-ATG13 can be phosphorylated and the SDS-PAGE band shifted up by the CDK1-CCNB complex in the in vitro kinase assay, and is antagonized by RO-3306, the CDK1 inhibitor. Furthermore, we examined the ULK1-ATG13 immunoprecipitates using CDK substrate phosphorylation motif antibodies in thymidine- and nocodazole-arrested cells throughout the cell cycle and confirmed ULK1 and ATG13 are both the direct substrates of CDK1 in mitosis and are differentially regulated during the cell cycle.
Figure 1. The model illustrates the roles of ULK1-ATG13 in the cell cycle and its phospho-regulation and function in mitosis. When cells are in mitosis, CDK1-CCNB/cyclin B phosphorylates ULK1-ATG13 to induce a significant electrophoretic bandshift. The phosphorylated ULK1-ATG13 promotes mitotic autophagy and cell cycle progression. In asynchronous conditions, double knockout of ULK1 and ATG13 inhibits cancer cell proliferation in both cell and mouse models. I, interphase; M, mitosis
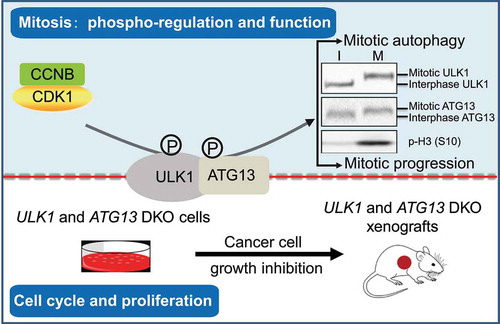
What is the function of ULK1-ATG13 hyperphosphorylation in mitosis? To answer this question, we combined Scansite prediction, mass spectrometry analysis, and site-directed mutagenesis, and identified 11 sites in ULK1 and 4 sites in ATG13. The alanine mutants significantly decreased the bandshift and the phospho-serine/threonine-proline signals. To unravel the function of phosphorylation events, we constructed ULK1 and ATG13 WT or alanine double-mutant cells in ULK1 and ATG13 DKO (double knockout) parental cells. However, these alanine mutations do not change ULK1-ATG13 localization or ULK1 kinase activity. Conversely, GFP-LC3-RFP reporter, LC3B-II turnover, and LC3B puncta number assays all showed that the autophagic flux decreases in ULK1 and ATG13 mutant cells, indicating that the mitotic ULK1-ATG13 hyperphosphorylation promotes mitotic autophagy.
Given that the ULK1-ATG13 level fluctuates in the cell cycle, do they function in cell cycle regulation? Our data show that ULK1-ATG13 functions as a complex to promote the cell cycle and cell proliferation. Using cell cycle synchronization and p-H3 (S10) staining, we found that ULK1 and/or ATG13 knockout (KO) delays mitotic entry. In addition, ULK1 and ATG13 DKO also inhibits cell cycle progression and cell proliferation, whereas ULK1 or ATG13 KO alone does not or only moderately affects them. Moreover, in nude mice bearing ulk1 and atg13 single-KO or DKO cells, the tumor weight and volume of the DKO group are significantly lower than all the other groups. In contrast, ULK1 inhibitor SBI-0206965 and/or atg13-KO alone are not strong enough to inhibit tumor growth as efficiently as the DKO, indicating that targeting ULK1 and ATG13 together might be a potential anti-cancer strategy.
Some important experimental details should be pointed out here, which are critical for investigating phosphorylation in mitosis. (1) The ULK1 antibody (Cell Signaling Technology, 8054) we originally used could detect the ULK1 bandshift only after the phosphate group was removed, which was probably caused by hyperphosphorylation-induced protein conformational change. However, another ULK1 antibody (Cell Signaling Technology, 4776/GeneTeX, GTX132669) and FLAG antibody (FLAG-ULK1) could both detect the bandshift. Therefore, we recommend using multiple antibodies to get a better judgment for hyperphosphorylated targets. (2) Mitotic slippage (cells exit mitosis prematurely without proper chromosome segregation and enter the next interphase) has occurred when the mitotic cells are treated with CDK1-AURKA inhibitors for 1.5 h, which abolishes the ULK1-ATG13 bandshift indirectly by mitotic slippage, but not the upstream kinase activity inhibition. In contrast, when using shorter timepoints (3 min-1 h), only the CDK1 inhibitor RO-3306 still can abolish the ULK1-ATG13 bandshift, but not the AURK inhibitors. Therefore, when studying kinase function in mitosis, the inhibitor treatment time should be carefully controlled to avoid mitotic slippage.
However, there are still some open questions. (1) The ULK1-ATG13-CDK1 interaction occurs in asynchronous and mitotic cells to a similar extent. Does CDK1 alone or with other cyclins regulate ULK1-ATG13 in interphase? (2) We found that ULK2 was also phosphorylated and the gel band shifted up in mitosis. Does ULK2 complement ULK1 in mitosis? We did ULK2 knockdown in ULK1-KO cells, which delays mitotic entry compared to ULK1-KO (data not shown). (3) Although we identified sites for ULK1-ATG13 bandshift, more work should be done to find out whether each phosphorylation site is functional, to identify the interactome of mitotic ULK1-ATG13, and to depict the phosphorylation landscape of all autophagy regulators in mitosis. Phosphatases that dephosphorylate ULK1-ATG13 in mitotic progression need to be confirmed. (4) Also, given the crosstalk between the ubiquitin-proteasome system and autophagy or mitosis, the ubiquitin modification and function of ULK1-ATG13 in mitosis will be an exciting direction for future investigation.
Finally, to address if CDK1 can overcome MTOR-AMPK signaling, we prepared alanine mutants of MTOR and AMPK phosphorylation sites in asynchronous cells, including ULK1-S757, ULK1-S637, ULK1-S555, ATG13-S224, and ATG13-S259. Whereas only ATG13S224A, an AMPK phosphorylation site mutant, affects bandshift, treatment with compound C does not, indicating that CDK1 might substitute for AMPK in mitosis to phosphorylate ATG13-S224. Besides, ATG13-S259 and ULK1-S757 (human S758), two previously reported phosphorylated sites of CDK1, do not affect the ATG13-ULK1 bandshift in mitosis, whereas their precise roles in mitosis need to be validated. Interestingly, from TCGA, some ULK1-ATG13 mutations in patients are associated with the phosphorylation motif/sites identified in our study, indicating some potential clinical significance of these phosphorylation sites that should be investigated in the future.
Disclosure statement
No potential conflicts of interest were disclosed.
Additional information
Funding
Reference
- Li Z, Tian X, Ji X, et al. ULK1-ATG13 and their mitotic phospho-regulation by CDK1 connect autophagy to cell cycle. PLoS Biol. 2020;18(6):e3000288.