
ABSTRACT
Macroautophagy/autophagy can selectively degrade misfolded proteins, damaged organelles and other cargoes. It is conceivable that alteration of the degradation processes could disrupt normal cellular signaling and contribute to human diseases such as cancer. To explore the link between aberrant autophagy selectivity and human cancer, we have developed a pipeline called “inference of cancer-associated LC3-interacting region-containing proteins” (iCAL), which integrates a sequence-based predictor, a model-based computational method, publicly available cancer mutations, and multiple experimental approaches. Using iCAL, we have identified 222 LIR motif-associated mutations (LAMs) in 148 LIR-containing proteins (LIRCPs), and validated that LAMs in ATG4B, STBD1, EHMT2 and BRAF impair their interactions with LC3 and/or autophagy activities. Moreover, we uncovered that STBD1, a previously poorly-characterized protein, inhibits tumor growth via metabolism reprogramming in cancer cells. A patient-derived mutation in STBD1 (W203C) disrupts the interaction with LC3 and promotes tumor growth. Taken together, iCAL provides an exciting new avenue to discover novel autophagy pathways that contribute to carcinogenesis.
Autophagy is an evolutionarily conserved catabolic process in eukaryotes that is essential for maintaining the homeostasis of cells, tissues and organisms through the degradation of misfolded proteins, damaged organelles, senescent cells and pathogens. Cargoes are selectively targeted for degradation via multiple mechanisms, including the LC3-interacting region (LIR). LIR-containing proteins (LIRCPs) function in every stage of autophagy, including the initiation and growth of phagophores (the precursors to autophagosomes), cargo recognition, transport, and autophagosome-lysosome fusion. The connection between autophagy and cancer is well-established. Aberrant regulation of autophagy can contribute to tumorigenesis and metastasis. However, it is less clear whether alternations in cancer genomes contribute to cancer development via altering autophagy selectivity. To address this question, we developed an integrated pipeline, iCAL, to investigate human cancer mutations in the LIR motif [Citation1] (). We show that iCAL furnishes a resource to discover new autophagy pathways for the development of cancer.
Figure 1. iCAL includes pLIRm (a new algorithm to predict the LIR motif), pLAM (a model-based algorithm to predict LIR motif-associated mutations), a pan-cancer analysis, and cell- and animal-based validations. We identified 148 LIR-containing proteins (LIRCPs) that carry single point mutations within the LIR motif, including some ATG genes, autophagy regulators and many novel candidate genes. Among these candidate genes, we further demonstrate that STBD1 possesses a previously unappreciated role in suppressing cancer growth. Transcriptomics, metabolomics and additional experimental assays were used to dissect the mechanism of STBD1 in tumor proliferation. 2-DG, 2-deoxyglucose
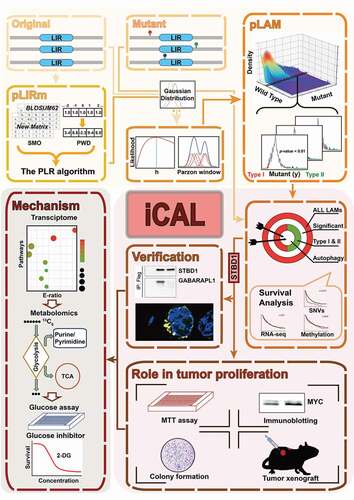
We first collected 127 experimentally verified LIR sequences in 105 LIRCPs, including 89 and 11 LIR motifs in Homo sapiens and Saccharomyces cerevisiae, respectively, for model training, and developed a new prediction tool named pLIRm. We show that pLIRm is more accurate than other existing methods, including iLIR, and hfAIM, in detecting the LIR motifs. We also collected cancer mutation data from multiple public databases including TCGA (The Cancer Genome Atlas), ICGA (International Cancer Genome Consortium) and COSMIC (Catalog of Somatic Mutations in Cancer), and obtained 2,963,952 non-redundant missense single nucleotide variants (SNVs). We then developed a model-based algorithm named pLAM to prioritize LAMs that could significantly change binding potentials to LC3. Using pLIRm, pLAM and cancer mutation data, we identified 222 potential LAMs in 148 LIRCPs, including 60 type I (increase the binding potential to LC3) and 162 type II (decrease the binding potential to LC3).
From 148 LIRCPs, we selected 5 of them (ATG4B, EHMT2, BRAF, ERCC6, and STBD1) for experimental validation. All of them, except for ERCC6, show alterations in LC3 binding and/or autophagy activities, as predicted by our algorithm. Remarkably, one LAM in BRAF (P453L) shows a gain of binding to LC3, although its functional relevance remains to be determined. These data suggest that our algorithm is useful to predict novel LIR motifs and those altered in cancer.
We next focused on STBD1 in our following studies for a few reasons. First, STBD1 was a known receptor in glycophagy (a process that degrades glycogen via the autophagic pathway) prior to our study, but its role in cancer is unknown. Second, the expression of STBD1 is significantly downregulated in multiple human cancers. Third, a predicted LAM in STBD1 (W203C) is present in an intestinal adenocarcinoma sample. Using multiple cell lines and a mouse tumor xenograft model, we found that overexpression of STBD1 inhibits the growth of cancer cells, whereas silencing of STBD1 is pro-tumorigenic. Remarkably, the patient-derived mutation W203C behaves similar to STBD1 knockdown in vitro and in vivo, indicating that STBD1 inhibits tumor growth through interacting with LC3B/ GABARAPL1.
We performed immunofluorescence experiments and glycogen content assays, and found that overexpression of STBD1W203C, but not WT STBD1, abolishes the colocalization of glycogen with GABARAPL1 and leads to the accumulation of glycogen in HCT116 cells. To understand how STBD1 inhibits tumor growth, we compared the gene expression profiles of shControl and shSTBD1 HCT116 cells as well as shSTBD1 HCT116 cells rescued with shRNA-resistant WT STBD1 (shSTBD1/WT) and STBD1W203C (shSTBD1/W203C). RNA-seq analysis, quantitative RT-PCR, and bioinformatics studies together demonstrated that the glycolysis-gluconeogenesis pathway is one of the most important pathways affected by STBD1; depletion of STBD1 leads to elevated expression of multiple enzymes responsible for glycolysis. The STBD1W203C mutation leads to a change of gene expression pattern, somewhat similar to STBD1 silencing. Thus, STBD1 inhibits tumor growth, likely through multiple mechanisms including altering gene transcription and reprograming the glycolysis pathway.
To further confirm the above results, we performed metabolomic profiling of shControl and shSTBD1 HCT116 cells. We found that STBD1 suppression enhances glycolysis, boosts the TCA cycle, and promotes nucleotide biosynthesis through the pentose phosphate pathway. Our metabolomic measurement is largely consistent with our observations in the transcriptomics, which included upregulation of multiple enzymes critical for the glycolytic pathway. As proliferating cells require more RNA and DNA synthesis, our results suggested that suppression of glycophagy could promote tumorigenesis via metabolic reprogramming. Guided by the above results, we further found that 2-deoxyglucose, a known glycolytic inhibitor, inhibits the proliferation of STBD1-suppressing cells more profoundly than that of control cells. Thus, inhibition of glycolysis could represent a personalized therapeutic approach for cancer patients with lower STBD1 expression level or expressing STBD1 mutants.
Overall, iCAL is a useful resource to investigate the link between aberrant autophagy and human cancer. Using iCAL, we have identify the missing link between glycophagy and tumor suppression. We hope that iCAL can help the community to discover novel autophagy pathways for cancer development, and lead, ultimately, to new cancer therapeutic approaches via manipulating these pathways.
Acknowledgments
Changjiang Scholars Program of China, and the program for HUST Academic Frontier Youth Team.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
Reference
- Han Z, Zhang W, Ning W, et al. Model-based analysis uncovers mutations altering autophagy selectivity in human cancer. Nat Commun. 2021;12(1):3258.