
ABSTRACT
Sjögren syndrome (SS) is a chronic and progressive autoimmune disease characterized by dry mouth and dry eyes, and characteristic autoantibodies. Evidence of altered macroautophagy/autophagy and apoptosis has been associated with SS, but a mechanistic understanding of the gene expression changes associated with these abnormal processes has not been realized. Recently, increased LAMP3 (lysosomal associated membrane protein 3) expression was found in a subset of SS patients and was associated with increased apoptosis and autoantigen accumulation and release. To better understand how LAMP3 expression might modulate apoptosis, cell biology, and biochemical studies were used to examine the effect of LAMP3 expression in minor salivary gland cells. LAMP3 expression resulted in degradation of LAMP1 increasing lysosomal membrane permeabilization and relocalization of cathepsins to the cytoplasm, resulting in destabilizing autophagic flux and caspase activation. These findings highlight the central role of LAMP3 expression in the pathogenesis of SS.
Abbreviations
A253-control: A253 control for LAMP3 stable overexpression; A253- LAMP3: A253 LAPM3 stable overexpression; CASP1: caspase 1; CASP3: caspase 3; CHX: cycloheximide; CTSB: cathepsin B; CTSD: cathepsin D; CQ: chloroquine; DCs: dendritic cells; ER: endoplasmic reticulum; LGALS3: galectin 3; HCV: hepatitis C virus; HSG-control: HSG control for LAMP3 stable overexpression; HSG-LAMP3: HSG LAMP3 stable overexpression; HSP: heat shock protein; HTLV-1: human T-lymphocyte leukemia virus-1; IXA: ixazomib; LAMP: lysosomal associated membrane protein; MHC: major histocompatibility complex; mAb: monoclonal antibody; OE: overexpression; pepA: pepstatin A; pAb: polyclonal antibody; pSS: primary Sjögren syndrome; qRT-PCR: quantitative real- time reverse transcriptase polymerase chain reaction; SLE: systemic lupus erythematosus; SS: Sjögren syndrome; UPR: unfolded protein response; V-ATPase: vacuolar-type proton- translocating ATPase; Y-VAD: Ac-YVAD-cmk; Z-DEVD; Z-DEVD-fmk; Z-VAD: Z-VAD- fmk
Introduction
Sjögren syndrome (SS) is a chronic autoimmune disease characterized by exocrine gland dysfunction (e.g. dry mouth and dry eyes) and lymphocytic infiltration of the affected organs (e.g. lymphocytic sialadenitis and dacryoadenitis) [Citation1,Citation2]. The loss of exocrine gland function in SS may relate in part to atrophy and fibrosis of the salivary and lacrimal parenchyma [Citation3,Citation4]. One mechanism explaining this phenomenon may be increased rates of apoptosis within the epithelium [Citation5]. Although apoptosis is long implicated in the development of SS [Citation6], recent studies have also demonstrated increased macroautophagy/autophagy markers in the lacrimal glands of SS patients and in CD4+ T-lymphocytes infiltrating the salivary glands, implicating a role for autophagy [Citation7,Citation8].
Autophagy is a dynamic cellular process initiated in response to cellular stressors such as endoplasmic reticulum (ER) stress [Citation9], and starvation [Citation10]. During autophagy, proteins, organelles and cytoplasm become enveloped within double-membrane vesicles called autophagosomes [Citation11]. After fusion with lysosomes, these cellular constituents are degraded [Citation11], and recycled to maintain cellular metabolic homeostasis [Citation11]. Autophagy plays a protective role and regulates cell death in cooperation with apoptosis.
Autophagy contributes to antigen presentation by major histocompatibility complex (MHC) class II in antigen-presenting cells, including dendritic cells (DCs), macrophages, and B cells via the degradation of internalized extracellular pathogens [Citation12]. Failure in steps of the autophagic process is associated with accumulation of misfolded intracellular proteins such as phosphorylated SNCA/α-synuclein [Citation13,Citation14] and amyloid-β [Citation15,Citation16] in Alzheimer and Parkinson diseases.
Due to its role in protein degradation and antigen presentation, autophagy has been implicated in the development of autoimmune diseases including rheumatoid arthritis [Citation12], psoriasis/psoriatic arthritis [Citation12], multiple sclerosis, inflammatory bowel disease [Citation12], and systemic lupus erythematosus (SLE) [Citation12]. Genome-wide association studies and variant identification studies have identified associations between genes in the autophagy pathway and the development of SLE [Citation17]. Marker genes associated with autophagy including BECN1/beclin1, MAP1LC3/LC3, and SQSTM1/p62 are reported to be differentially expressed in peripheral blood mononuclear cells from SLE patients compared with controls [Citation18]. Finally, mice lacking LC3-associated phagocytosis develop an SLE like phenotype [Citation19].
The lysosome is a membrane-bound organelle with acidic pH maintained by the vacuolar-type proton-translocating ATPase (V-ATPase), and is associated with cell death via lysosomal membrane permeabilization leading to lysosome-dependent cell death [Citation20,Citation21]. A key to this pathway is the tightly controlled function of the lysosome. Although the expression of LAMP1 (lysosomal associated membrane protein 1) and LAMP2 is often associated with protection from lysosomal membrane permeabilization [Citation22], the role of LAMP3 expression on lysosomal membrane permeabilization remains unclear. In recent studies, LAMP3 expression has been observed to be limited to specific cell types such as DCs [Citation23,Citation24] and type II pneumocytes in human [Citation23]. LAMP3 expression is induced by ATF4 induction [Citation25] following ER stress initiated by hypoxia [Citation26], irradiation [Citation27], and infection [Citation28]. LAMP3 is a canonical interferon stimulated gene specifically induced by viral infections (hepatitis C virus [HCV], human papilloma virus, influenza) [Citation29,Citation30]. Viruses have long been implicated in the pathogenesis of SS [Citation31,Citation32]. Interestingly, autoantibodies (anti-TRIM21/RO52, anti-RO60/TROVE2) associated with SS, are known to be induced by several chronic viral infections including HCV, and human T-lymphocyte leukemia virus-1 (HTLV1). These infections often involve the salivary glands and induce characteristic sicca symptoms, xerostomia (dry mouth) [Citation33–41]. We previously demonstrated a mechanism by which LAMP3 overexpression (OE) induces TRIM21 aggregation, relocalization, and autoantigen presentation [Citation42]. Increased LAMP3 expression is detected in the salivary glands of some SS patients and reported to be associated with apoptosis [Citation42]. Because of the intimate crosstalk between apoptosis and autophagy, it is possible that LAMP3 expression may induce apoptotic cell death via alterations in autophagy.
In this study, human salivary gland biopsies and in vitro models were used to investigate the effect of LAMP3 expression on autophagy. Specifically, LAMP3 OE obstructed autophagy at a step after autophagosome formation via degradation of LAMP1. This altered protein behavior led to the inappropriate release of CTSB (cathepsin B) and CTSD (cathepsin D) into the cytoplasm resulting in apoptosis via activation of CASP1 (caspase 1) and BID-CASP3 (caspase 3) pathways. Our findings highlight a novel crosstalk between apoptosis and autophagy via altered lysosome membrane integrity. This mechanism may be central to the cell death associated with SS.
Results
LAMP3 expression correlates with the number of LC3 puncta in minor salivary glands of SS patients
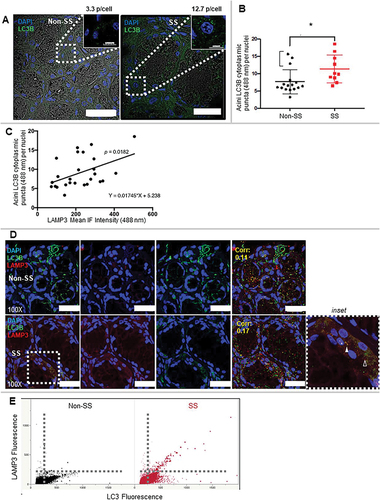
An altered number of LC3 aggregates or puncta is an important marker of autophagy [Citation43]. In order to investigate the relationship between LAMP3 expression and autophagy in SS, confocal images of serial sections of minor salivary glands from control nonautoimmune sicca subjects or SS patients were probed with anti-LC3B or LAMP3 antibodies. SS patients exhibited an increased number of LC3B puncta in minor salivary glands compared with the control subjects (). Despite the statistically significant increase in LC3B puncta per cell in SS subjects minor salivary glands, three non-SS salivary glands exhibited high levels of LAMP3, similar to SS. In 2/3 individuals, subjects had autoantibodies to Ro/SSA but failed to meet 2002 American-European Consensus Group criteria [Citation44] (a single patient met 2016 American College of Rheumatology criteria [Citation45] but not American-European Consensus Group 2002 criteria). Previous research has reported a positive association between LAMP3 expression and autoantibody formation [Citation42]. Due to the similar expression and subcellular localization patterns of LC3B in SS, the relationship between LAMP3 expression and LC3B expression was compared using a correlation analysis. A positive correlation between LAMP3 expression and LC3B puncta number per cell was found (). We performed confocal immunofluorescence to determine the subcellular colocalization of LC3B with LAMP3 using an automated spot detector and colocalization algorithms (spot detector, colocalization suite, icy) (). Unexpectedly, we found increased LC3B puncta number and increased per vesicle expression of LAMP3 in SS epithelial cells, and increased colocalization of the LC3B and LAMP3 signals. Moreover, larger-sized puncta were more commonly LAMP3 positive, as illustrated by (). These findings suggest a potential link between LAMP3 expression and autophagic flux. Alternatively, LAMP3 expression could directly impact the size and number of colocalized LC3 puncta.
Figure 1. SS patients have a positive correlation between LAMP3 expression and LC3B puncta. (A, B) LC3B staining in the same representative cases. LC3B highlights the acinar, ducts, and lymphocytic infiltrates in SS more so than Non-SS including abundant perinuclear LC3 aggregates in acini of the SS case. Scale bar: 30 μm. Using ICY image analysis, LC3B puncta (0.5–2.0 μm) were counted using a spot detection algorithm and compared relative to the total number of nuclei per acini. Three representative images with at least 3–5 acini per image at 1000X resolution with a 60 z-stack series of images taken at 0.2 μm resolution (x,y,z) were counted and compared between non-SS and SS. Isotype controls for each case demonstrated negligible nonspecific staining (data not shown). The outlier non-SS (bracketed) cases with numbers of LC3B puncta similar to SS had clinical profiles suggestive of SS but did not strictly meet 2002 AECG classification criteria including: 2/3 had autoantibodies to Ro/SSA and also either subjective or objective dry mouth and/or dry eyes, but not both; or incomplete clinical data for a single subject. These results indicate possible evolving SS or other autoimmune disease and also the clinical heterogeneity of SS. Because of our strict adherence to a priori classifications of subjects in this data set, the cases were retained with this caveat. (C) Analysis of correlation of LAMP3 expression and the number of LC3B puncta was performed with Pearson correlation. (Corr: 0.221, p = 0.0182; Pearson’s correlation). (D, E) LAMP3 and LC3B staining in the same representative cases. Scale bar: 25 μm. LAMP3 and LC3B levels in each puncta were analyzed and then plotted. The size of each dot in the graph means their relative size. * p < 0.05, Student’s t-test.
Both LAMP3 expression and autophagic flux are reported to be controlled by the unfolded protein response (UPR) initiated by ER stress [Citation25–28,Citation46]. To study whether ER stress correlates with LAMP3 expression and LC3B puncta in SS, the expression of genes associated with UPR was analyzed. Although normalized mRNA expression across 13 primary SS patients and 15 healthy controls did not show a clear relationship with UPR gene expression (Figure S1A), further analysis identified a subset of SS patients that exhibited a significant elevation in LAMP3, as well as other ER stress associated genes including UBD, KLHL6, ATF3, DERL3, BATF, BIRC3, and CREB3L1 (Figure S1B). These findings suggested that ER stress could be increased in minor salivary glands in a subset of SS patients.
LAMP3 expression results in a reduction of autophagic flux
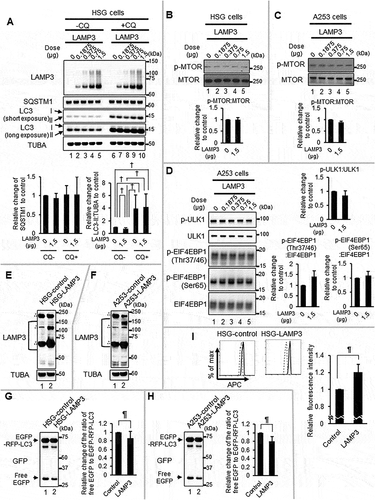
To study whether LAMP3 had an effect on autophagy, LAMP3 was overexpressed in HSG and A253 cells. The effect of LAMP3 on autophagy was investigated by quantifying the ratio of LC3-II to TUBA/α-tubulin along with SQSTM1 expression in control and LAMP3-OE cells of HSG and A253 cells. The ratio of LC3-II to TUBA in LAMP3-OE cells treated with chloroquine (CQ) was increased similar to control cells treated with CQ, while LAMP3 OE alone induced a decrease in the ratio of LC3-II to TUBA ( and S2A) suggesting LAMP3 expression could induce a change in autophagic flux.
Figure 2. LAMP3 decreased protein degradation via inhibited autophagy. (A) Representative blotting of HSG control and LAMP3-OE cells treated with or without 25 μM CQ for 6 h. The ratio of LC3-II to TUBA and SQSTM1 to TUBA was calculated, and relative change to control was plotted. Values shown are mean ± SD. SQSTM1: N = 4; LC3-II: N = 7. The effect of LAMP3 on phosphorylation of MTOR in (B) HSG and (C) A253 cells was analyzed using western blot and representative blotting was shown. The ratio of phosphorylated MTOR (p-MTOR) to total MTOR was calculated, and relative change to control was plotted. The values shown are mean ± SD. N = 5 (HSG cells); N = 4 (A253 cells). (D) Representative blots show total and phosphorylated protein of ULK1 and EIF4EBP1. The ratio of phosphorylated protein to total protein in each protein was calculated, and relative change to control was plotted. Values shown are mean ± SD. N = 3. Representative blotting of (E) HSG and (F) A253 LAMP3 stable OE cells (HSG-LAMP3 cells and A253-LAMP3 cells). “∴” indicates nonspecific band. The ratio of EGFP-RFP-LC3 protein degradation in (G) HSG-control and HSG-LAMP3 cells, and (H) A253-control and A253-LAMP3 cells was examined using western blot and representative blotting was shown. The ratio of free EGFP to EGFP-RFP-LC3 was calculated, and relative change to control was plotted. Values shown are mean ± SD. N = 10 (HSG cells); N = 4 (A253 cells). (I) The long-lived protein degradation assay was performed as described in materials and methods (Dashed line: without labeling; Bold line: with labeling). The relative change of fluorescence intensity to HSG-control cells was plotted. Values shown are mean ± SD. N = 5. ¶ p < 0.05, Ratio paired t-test. † p < 0.05, †† p < 0.01, ††† p < 0.001, Tukey’s multiple comparisons test.
CQ treatment can increase the number of LC3 puncta as a result of inhibiting the progression of autophagy [Citation47]. Although an increase in LC3B puncta was observed in HSG cells with CQ treatment or LAMP3 expression, the increase was not statistically significant. However, CQ treatment and LAMP3 expression did significantly increase LC3B puncta size. In the LAMP3-OE cells, the increase was independent of CQ treatment (Figure S2B–D). These findings also support the idea that autophagic flux was altered in the LAMP3-OE cells.
Protein SQSTM1, one of the receptors in different types of selective autophagy, is reported to have a variable response in association with autophagic degradation [Citation48]. However, no significant difference in SQSTM1 expression was found between control and LAMP3-OE cells of HSG and A253 cells treated with or without CQ (, S2A). Because MTOR and its downstream substrates regulate autophagic flux, the ratio of phosphorylation of MTOR and its substrates were compared in control and LAMP3-OE cells. No differences in the ratio of p-MTOR to MTOR was found in LAMP3-OE HSG and A253 cells (). Moreover, no significant difference of the phosphorylation of MTOR substrates ULK1, or p-EIF4EBP1 (Thr37/46, Ser 65) were found in A253 control and LAMP3-OE cells ().
Based on these results, we explored the possibility that LAMP3 inhibited autophagy at a downstream step after complete closure of the autophagosome. To test this hypothesis, HSG LAMP3 stable OE cells (HSG-LAMP3 cells) and A253 LAMP3 stable OE cells (A253-LAMP3 cells) were established by transfection of LAMP3 encoding plasmid, selected (), and then used to study protein degradation by monitoring changes in EGFP-RFP-LC3 protein as a reporter of protein degradation. Fusion of the reporter protein-containing autophagosome with the lysosome generates an autolysosome, which mediates proteolysis of the fusion protein, as well as that of the endogenous LC3. Even though LC3 is degraded, the free EGFP moiety released during degradation is relatively resistant to proteolysis, and the accumulated EGFP is easily monitored. Hence, the ratio of free EGFP to EGFP-RFP-LC3 can be used to monitor autophagic flux [Citation49,Citation50]. Interestingly, the ratio of free EGFP to EGFP-RFP-LC3 was significantly decreased in HSG-LAMP3 and A253-LAMP3 cells compared with control cells (). To confirm that LAMP3 OE reduced autophagic protein degradation, the rate of protein degradation was monitored using azidohomoalanine labeling in a long-lived protein degradation assay in HSG-LAMP3 cells [Citation51]. In agreement with the results of the EGFP-RFP-LC3 degradation experiment, the fluorescent intensity in azidohomoalanine labeled HSG-LAMP3 cells was significantly elevated relative to control cells (). These results suggest that LAMP3 was able to inhibit autophagy-mediated protein degradation at a step after autophagosome formation.
LAMP3 expression leads to lysosomal dysfunction by destabilizing the lysosomal membrane
Autophagosome fusion with the lysosome initiates autophagic protein degradation by introducing lysosomal enzymes into the autophagosome and acidifying the luminal contents of the autolysosome [Citation52]. Confocal imaging of the lysosomal marker, LAMP1 and LC3 in LAMP3 transfected cells or control LAMP3-negative cells, showed little LAMP1-LC3 colocalization in the LAMP3-OE cells, compared with control cells (Figure S2B,E). These results suggest that LAMP3 could have an effect on the formation of the autolysosomes by inhibiting fusion or by decreasing the lysosome as a result of the decrease in LAMP1.
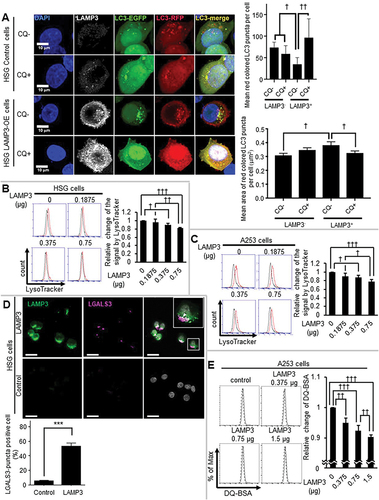
Tandem constructs such as EGFP-RFP-LC3 and EGFP-mCherry-LC3 are useful in distinguishing between autophagosomes and autolysosomes [Citation48,Citation53]. Under neutral pH conditions, LC3 puncta containing RFP and EGFP reporter proteins are simultaneously fluorescent, resulting in colocalized red and green fluorescence (yellow color). Under acid conditions (e.g. autolysosome), EGFP protein fluorescence is quenched resulting in differential RFP fluorescence (red colored LC3 puncta). Confocal imaging of HSG LAMP3-positive and LAMP3-negative cells with or without CQ, exhibited that LAMP3-negative LC3 puncta, which are the majority of LC3 puncta in HSG LAMP3-OE cells, were yellow similar to LC3 puncta found in control cells treated with CQ (). Moreover, quantification of red-colored LC3 puncta indicated a decrease in their number in LAMP3-OE cells with an increase in their area (). Flow cytometry analysis using LysoTracker indicated a significant decrease in lysosome acidification in LAMP3-OE cells () suggesting that LAMP3 expression induced lysosomal instability. To further investigate whether LAMP3 expression impaired the protein degradation activity of the lysosome, LAMP3 expressing cells were treated with DQ-BSA and monitored for a change in fluorescent intensity compared with control cells. Treatment of LAMP3-OE A253 cells showed a decrease in fluorescence compared with control cells suggesting LAMP3 expression induced a decrease in lysosomal degradation (). Moreover, HSG LAMP3-OE cells showed an increase in LGALS3 (galectin 3) accumulation (), indicative of damage to the lysosomal membrane. These results suggest that LAMP3 expression decreased lysosomal protein degradation activity due to compromised lysosomal membrane integrity.
Figure 3. LAMP3 induced dysfunction in the lysosome. (A) The acidic environment in HSG control and LAMP3-OE cells was analyzed using EGFP-RFP-LC3 protein with or without CQ treatment. The number and mean area of red colored LC3 puncta in each cell were analyzed and plotted. GFP-positive cells in the cells co-transfected with 0.75 μg of GFP plasmid and a total 0.75 μg of pME18S-empty and pME18S-LAMP3 plasmid of (B) HSG cells and (C) A253 cells were analyzed using Flow cytometry with LysoTracker DND-99 (Black line: without LysoTracker; Red line: with LysoTracker). The mean fluorescence intensities compared with control were shown. Values shown are mean ± SD. N = 3. (D) Immunocytochemistry analysis of HSG control (lower panels) and LAMP3-OE cells (upper panels) was performed. Scale bar: 50 μm. LGALS3-puncta positive cells (in 100 of control or LAMP3-OE cells) were counted, and the percentage was plotted. Values shown are mean ± SD. N = 4. (E) Lysosomal protein degradation capacity in A253 control and LAMP3-OE cells were measured using DQ-BSA (Dashed line: without DQ-BSA; Normal line: with DQ-BSA). Relative changes to control were plotted. Values shown are mean ± SD. N = 4. *** p < 0.001, unpaired Student’s t-test. † p < 0.05, †† p < 0.01, ††† p < 0.001, Tukey’s multiple comparisons test.
LAMP3 expression induces LAMP1 protein degradation
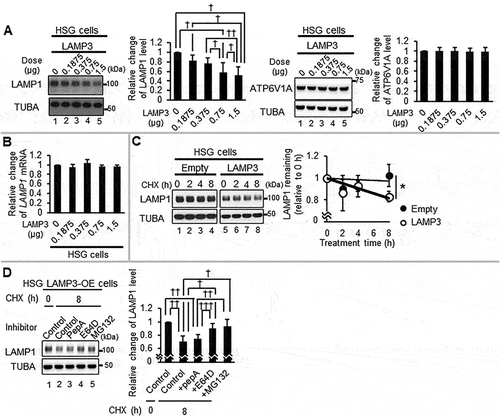
LAMP1 expression is required for lysosomal integrity and loss of LAMP1 expression is associated with lysosomal membrane permeabilization [Citation22]. Although no significant difference was observed in LAMP1 mRNA levels in HSG control and LAMP3-OE cells, western blotting showed a decrease in LAMP1 protein level in HSG LAMP3-OE cells compared with control empty plasmid transfected cells (). In contrast, no significant difference in the lysosomal protein ATP6V1A was observed in HSG LAMP3-OE cells compared with HSG control cells (). To investigate whether the decrease in LAMP1 was dependent on posttranscriptional protein degradation, LAMP1 protein levels following cycloheximide (CHX) treatment were compared in HSG control and LAMP3-OE cells. A significant decrease in LAMP1 expression was detected in LAMP3-OE cells treated with CHX over 8 h compared with controls, suggesting a significant increase in post-transcriptional protein degradation of LAMP1 (). Similar results were found in A253 cells (Figure S3A). To characterize the mechanism of LAMP1 protein degradation, the decrease in LAMP1 protein was measured in HSG LAMP3-OE cells treated with CHX in combination with proteasome inhibitor, MG132, or the cathepsin inhibitors pepstatin A (pepA) and E64D. Treatment with MG132 or CTSB inhibitor, E64D, inhibited LAMP1 degradation compared to HSG LAMP3-OE control cells (). MG132 is reported to inhibit calpain and CTSB as well as the proteasome [Citation54–56]. To distinguish between these three mechanisms of LAMP1 degradation, specific proteasome inhibitors, VR23 and ixazomib (IXA), were also tested and showed no inhibition in LAMP1 protein degradation compared with HSG LAMP3-OE control cells (Figure S3B). These results suggest that LAMP3-associated LAMP1 protein degradation was dependent on CTSB activity. This increased degradation was likely the result of the release or increased expression of cellular proteinases associated with the lysosome. However, no significant differences in CTSD mRNA expression and CTSD protein levels were observed in HSG LAMP3-OE cells compared with controls (Figure S3C,D) supporting the increased degradation activity was the result of released lysosomal proteinases.
Figure 4. LAMP3 induced CTSB dependent degradation of LAMP1. (A) The expression of LAMP1 and ATP6V1A in HSG control and LAMP3-OE cells was examined using western blot and representative blotting was shown. The results were normalized by TUBA, and relative change to control was plotted. Values shown are mean ± SD. LAMP1: N = 3; ATP6V1A: N = 5. (B) LAMP1 mRNA expression levels in HSG control and LAMP3-OE cells were measured using qRT-PCR. N = 3. (C) HSG control and LAMP3-OE cells were incubated with 50 μg/ml CHX and harvested at 0, 2, 4 and 8 h. Cells lysates were analyzed by western blot. The amount of LAMP1 remaining after CHX treatment for 2, 4 and 8 h was calculated as the fold change of the protein present at 0 h. Values shown are mean ± SD. N = 3. (D) HSG control and LAMP3-OE cells were incubated with 50 μg/ml CHX following pretreatment with 10 μM pepA, E64D or MG132 for 1 h, and harvested at 8 h. Cells lysates were analyzed by western blot. The amount of LAMP1 remaining after CHX treatment for 8 h was calculated as the relative change of the protein to HSG LAMP3-OE control cells. Values shown are mean ± SD. N = 7. * p < 0.05, unpaired Student’s t-test. † p < 0.05, †† p < 0.01, ††† p < 0.001, Tukey’s multiple comparisons test.
LAMP3 induced relocalization of CTSB and CTSD to the cytoplasm
Lysosomal membrane permeabilization is often associated with a decrease in LAMP1 and the release of lysosomal cathepsins like CTSB and CTSD into the cytoplasm [Citation22,Citation57,Citation58]. To measure the subcellular localization of cathepsins (CTSB and CTSD), cell lysates were fractionated into cytoplasm and membrane-bound organelle fraction containing the lysosome. The composition of each fraction was confirmed by Western blots probed for the cytoplasmic marker TUBA, the membranous organelle marker ATP1A1/Na-K ATPase α1, and the lysosomal marker LAMP1. Additional western blots of CTSB and CTSD showed a significant increase in cathepsins in the cytoplasm of HSG LAMP3-OE cells compared with control cells (). Furthermore, the cytoplasmic CTSD activity in HSG LAMP3-OE cells was also significantly increased compared with controls (). These results suggest that LAMP3 expression resulted in the release of CTSB and CTSD from the lysosome into the cytoplasm.
Figure 5. LAMP3 induced relocalization of lysosomal enzymes to the cytoplasm. (A) Representative western blots of cytoplasmic LAMP1, CTSB and CTSD in HSG control and LAMP3-OE cells. Membrane-bound organelle fraction contained lysosomes and other organelles. Protein levels were normalized to TUBA, and relative protein levels to control were plotted. Values shown are mean ± SD. LAMP1: N = 5; CTSB: N = 4; CTSD: N = 3. Immature type of cathepsins, and mature type of cathepsins were indicated as “IM” and “M”, respectively. (B, C) CTSD activity in cytoplasm was measured as described in Materials and Methods. The obtained fluorescence intensity was normalized to sample protein concentration, and relative change to control was plotted. Values shown are mean ± SD. N = 3. (D) Confocal immunofluorescence analysis of HSG control and LAMP3 cells treated with or without CQ was performed. The correlation of LAMP1 and activated CTSD vesicles in each cell was investigated. Dotted or dashed squares signifies area of inset in row of images following subsequently for LAMP3+ and LAMP3− cells. Dotted and dashed ellipses are areas of LAMP3+ and LAMP3− cells demonstrating differential colocation of activated CTSD and LAMP1. (E) Confocal immunofluorescence analysis of HSG control and LAMP3 cells was performed. The correlation of V-ATPase (ATP6V1A) and activated CTSD vesicles in each cell was investigated. Dotted or dashed squares signifies area of inset in row of images for LAMP3+ and LAMP3− cells. Scale bar: 20 μm and 5 μm (inset). Dotted and dashed circles are areas of LAMP3+ and LAMP3− cells demonstrating differential colocation of activated CTSD and V-ATPase. ¶ p < 0.05, Ratio paired t-test. * p < 0.05, *** p < 0.001 unpaired Student’s t-test. †† p < 0.01, ††† p < 0.001, Tukey’s multiple comparisons test.
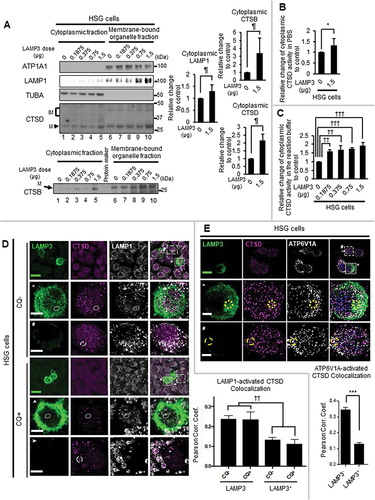
Altered localization of CTSD was confirmed by confocal imaging of LAMP1 and CTSD in LAMP3-OE HSG cells compared with controls. There was a statistically significant decrease in the colocalization of LAMP1 and activated CTSD in HSG LAMP3-OE cells compared with control cells (). Since this decrease in the colocalization of LAMP1 and activated CTSD could be induced by the decrease in LAMP1 protein shown in , the colocalization of lysosome and activated CTSD was further analyzed using V-ATPase, which showed no difference in expression in control and LAMP3-OE cells (). HSG LAMP3-OE cells exhibited a decrease in the colocalization of V-ATPase and activated CTSD compared to control cells (). These results support LAMP3-induced relocalization of lysosomal enzyme into the cytoplasm.
In addition to LAMP1 protein, a decrease in HSPA/HSP72 (heat shock protein family A (Hsp70)) is associated with a loss in lysosomal membrane integrity [Citation59] and release of lysosomal cathepsins into cytoplasm [Citation60]. In contrast to the LAMP3-dependent decrease in LAMP1, no decrease of HSPA protein levels was observed in HSG LAMP3-OE or A253 LAMP3-OE cells (Figure S3E,F).
A recent publication reported the release of lysosomal glycoprotein, LAMP1 and LAMP2 as a lysosomal membrane protein complex into the cytoplasm following lysosomal membrane permeabilization by L-leucyl-L-leucine methyl ester [Citation61]. Similarly, LAMP3 expression increased LAMP1 protein in the cytoplasmic fraction ().
These results suggest that LAMP3 could induce lysosomal membrane permeabilization via degradation of LAMP1 resulting in the release of CTSB and CTSD into the cytoplasm.
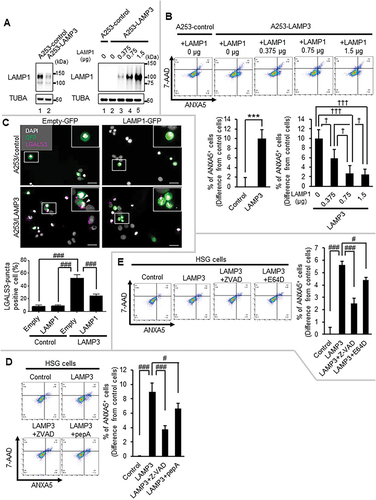
LAMP3 induced apoptosis via a cathepsin-dependent pathway
LAMP3 expression can induce apoptotic cell death [Citation42]. The above results suggest that LAMP1 protein degradation-induced lysosomal membrane permeabilization led to increased cytoplasmic CTSB and CTSD, resulting in apoptosis. To further study the effect of LAMP1 on LAMP3-induced apoptosis, LAMP1 protein degradation was complemented by the transfection of LAMP1 plasmid. LAMP1 OE prevented apoptotic cell death by LAMP3 in a dose-dependent manner (). To further study the effect of LAMP1 OE on LAMP3 induced lysosomal membrane permeabilization, the damage in lysosomal membrane was compared using LGALS3 puncta assay in A253-LAMP3 cells transfected with control or LAMP1 encoding plasmids. The transfection of a LAMP1-encoding plasmid into A253-LAMP3 cells showed a decrease in LGALS3-puncta positive cells compared with control plasmid transfected A253-LAMP3 cells (). These results suggest that a decrease in LAMP1 protein was required for LAMP3-induced apoptosis and highlights the importance of lysosomal homeostasis. LAMP3 induced the relocalization of lysosomal enzyme, cathepsins into the cytoplasm (). To examine the effect of cytoplasmic cathepsins on LAMP3-induced apoptosis, HSG LAMP3-OE cells were treated with CTSD inhibitor, pepA, and CTSB inhibitor, E64D. PepA treatment decreased apoptosis in HSG LAMP3-OE cells (). E64D treatment also decreased the apoptosis in HSG LAMP3-OE cells (). These results suggest that cytoplasmic CTSB and CTSD contributed to LAMP3-induced apoptosis.
Figure 6. LAMP3 induced apoptosis was inhibited by LAMP1 OE or the inhibitor of CTSB or CTSD. (A) Representative western blots of A253-LAMP3 cells transfected with GFP plasmid and LAMP1 plasmid or control plasmid. (B) Forty-eight hours post-transfection, apoptotic cells in GFP-positive cells were determined by Flow cytometry using APC-ANXA5/annexin V-7-AAD. The difference of the rate of ANXA5+ cells in GFP-positive cells of A253-LAMP3 transfected with LAMP1 cells from that in GFP-positive cells in A253-control cells was plotted. N = 4. (C) Immunocytochemistry analysis of A253-control (upper panels) and A253-LAMP3 cells (lower panels) transfected with empty-GFP plasmid (left side) or LAMP1-GFP plasmid (right side) was performed. Scale bar: 50 μm. LGALS3-puncta positive cells in 100 cells of each group were counted, and the percentage was plotted. Values shown are mean ± SD. N = 3. (D) HSG control and LAMP3-OE cells were incubated with or without 20 μM Z-VAD or 5 μM pepA for 14 h. Apoptotic cells in GFP-positive cells were determined by Flow cytometry using APC-ANXA5/7-AAD. The difference of the rate of ANXA5+ cells in GFP-positive cells of LAMP3-OE cells treated with or without 20 μM Z-VAD or 5 μM pepA from that in GFP-positive cells in control cells was plotted. Values shown are mean ± SD. N = 3. (E) HSG control and LAMP3-OE cells were incubated with or without 20 μM Z-VAD or 2.5 μM E64D for 14 h. Apoptotic cells in GFP-positive cells were determined by Flow cytometry using APC-ANXA5/7-AAD. The difference of the rate of ANXA5+ cells in GFP-positive cells of LAMP3-OE cells treated with or without 20 μM Z-VAD or 2.5 μM E64D from that in GFP-positive cells in control cells was plotted. N = 3. *** p < 0.001, unpaired Student’s t-test. † p < 0.05, ††† p < 0.001, Tukey’s multiple comparisons test. # p < 0.05, ### p < 0.001, Dunnett’s multiple comparisons test.
LAMP3 induced caspase-dependent cell death
Caspases play a central role in initiating cell death [Citation62,Citation63] and they can be activated by CTSB [Citation64] and CTSD [Citation65]. BID is activated by cleavage to tBID following enzymatic cleavage by endo-proteases such as CTSD, which then can transactivate CASP3 [Citation65]. No difference in the BID mRNA expression levels in HSG LAMP3-OE cells was found (). Moreover, a previous study showed that LAMP3 has no effect on CASP3 mRNA expression level [Citation42]. However, the protein level of BID and CASP3 in HSG LAMP3-OE cells was significantly decreased suggesting that they were cleaved to their activated forms (). In agreement, cleaved CASP3 activity in HSG LAMP3-OE cells was also elevated () and the LAMP3-induced cleavage of CASP3 could be inhibited by pepA treatment (), suggesting that LAMP3-induced activation of BID and CASP3 was via CTSD.
Figure 7. LAMP3 induced apoptosis is associated with the activity of CASP1 and CASP3. (A) BID mRNA expression levels in HSG control and LAMP3-OE cells were determined by using qRT-PCR. N = 3. (B) Representative western blots of HSG control and LAMP3-OE cells were performed to measure BID and CASP3 expression. BID and CASP3 expression levels were normalized to TUBA. The relative change compared with control was plotted. Values shown are mean ± SD. CASP3: N = 5; BID: N = 3. (C) Cleaved CASP3 activity in HSG control and LAMP3-OE cells was measured as described in Materials and Methods. The relative change compared with control was plotted. Values shown are mean ± SD. N = 3. (D) Fourteen hours post-treatment with 5 μM pepA, cleaved CASP3 activity was compared. The relative change to control was plotted. Values shown are mean ± SD. N = 4. (E) HSG control and LAMP3-OE cells were incubated with or without 20 μM Z-VAD or 10 μM Z-DEVD for 14 h. Apoptotic cells in GFP-positive cells were determined by Flow cytometry using APC- ANXA5/7-AAD. The difference of the rate of ANXA5+ cells in GFP-positive cells of LAMP3-OE cells treated with or without 20 μM Z-VAD or 10 μM Z-DEVD from that in GFP-positive cells in control cells was plotted. Values shown are mean ± SD. N = 4. (F) HSG control and LAMP3-OE cells were incubated with or without 20 μM Z-VAD or 50 μM Y-VAD for 14 h. Apoptotic cells in GFP-positive cells were determined by Flow cytometry using APC- ANXA5/7-AAD. The difference of the rate of ANXA5+ cells in GFP-positive cells of LAMP3-OE cells treated with or without 20 μM Z-VAD or 50 μM Y-VAD from that in GFP-positive cells in control cells was plotted. N = 4. ¶ p < 0.05, Ratio paired t-test. † p < 0.05, †† p < 0.01, ††† p < 0.001, Tukey’s multiple comparisons test. # p < 0.05, ### p < 0.001, Dunnett’s multiple comparisons test.
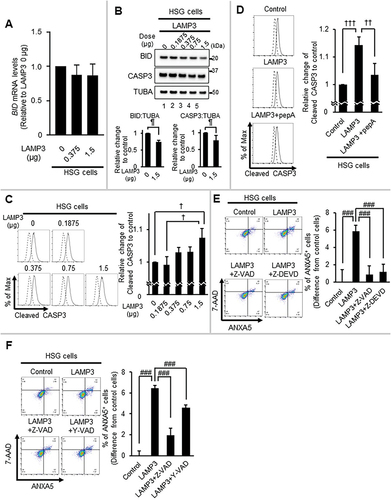
To investigate the role of CASP3 in LAMP3-induced apoptosis, HSG LAMP3-OE cells were treated with the CASP3-specific inhibitor, Z-DEVD-fmk (Z-DEVD). Z-DEVD treatment inhibited apoptosis in HSG LAMP3-OE cells (). Similarly, the cytoplasmic CTSB may contribute to cell death via CASP1 activation [Citation66,Citation67]. When HSG LAMP3-OE cells were treated with the CASP1-specific inhibitor, Ac-YVAD-cmk (Y-VAD), apoptosis was also decreased (). These results suggest that LAMP3 induced an increase in activation of both CASP3 and CASP1 pathways, resulting in apoptotic cell death.
Discussion
Although increased cell death via apoptosis is well established in SS [Citation6], little is known about the molecular mechanisms involved in this process. Increased LAMP3 expression has been found in several microarray transcriptomic studies of the minor salivary glands from SS patients [Citation68–70]. We recently reported that elevated LAMP3 expression increases apoptosis and is associated with the presence of autoantibodies in SS patients [Citation42] and that ectopic expression of LAMP3 in the salivary glands of mice induces an SS-like phenotype [Citation71]. In the present study, the relationship between LAMP3-induced apoptosis and LAMP3’s effect on the lysosome was analyzed in detail. One insight from pathway analysis of transcriptomic data of the minor salivary glands of SS patients was that SS patients broadly exhibited activation of the cellular stress response pathway (Figure S1A,B). Confocal imaging of minor salivary glands confirmed the correlation between LC3B accumulation, an important response to cell stress and a marker of autophagy, and LAMP3 expression. This observation, combined with in vitro experiments, confirmed LAMP3 expression inhibited autophagic flux. The change in the lysosomal compartment in LAMP3-OE cells also resulted in increased cytoplasmic CTSB and CTSD leading to activation of BID and CASP3, as well as CSAP1 and commitment to apoptosis. Importantly, the deleterious effects of LAMP3 could be blocked by LAMP1 OE or treatment with cathepsin inhibitors.
Autophagy is a dynamic protein degradation process that is initiated with the fusion of the autophagosome and the lysosome. The autophagic flux is monitored by the ratio of LC3-II to internal controls such as TUBA and ACTB/β-actin, the number of LC3 puncta in a cell, changes in SQSTM1 protein levels, and autophagy-dependent protein degradation [Citation43,Citation72]. As shown in , the number of LC3B puncta was increased in pSS patients and correlated with LAMP3 expression. Moreover, LAMP3 expression resulted in reduced degradation activity in both the EGFP-RFP-LC3 protein and long-lived protein degradation assays (). Although LAMP3-induced loss of LC3 resembled the activation of autophagic degradation of LC3, no additional accumulation of LC3-II upon LAMP3 OE in the presence of CQ was observed ( and S2A). Moreover, no decrease in protein levels of SQSTM1 upon LAMP3 OE was observed ( and S2A) suggesting that LAMP3 inhibited autophagy. Autophagy inhibition induced by autophagy-lysosome pathway inhibitors, CQ or bafilomycin A1, is reported to increase the release of autophagosome components such as LC3 in extracellular vesicles [Citation73]. In our hands, LAMP3 OE decreased LC3 in the absence of CQ ( and S2A) suggesting that LAMP3 could induce the release of LC3 via extracellular vesicles due to LAMP3-induced autophagy inhibition.
CQ, a lysosomotropic reagent, is reported to increase the size of LC3 puncta and inhibit autophagy flux. This process is done through the induction of LC3-II recruitment onto the endolysosomal vacuolar membrane following an osmotic imbalance induced by the accumulation of CQ in the endolysosomal compartment [Citation74–76]. Surprisingly, the size of LC3 puncta was increased upon LAMP3 OE similar to CQ (Figure S2B,D). LAMP3 is reported to exist on the lysosomal membrane, suggesting that LAMP3 could also induce an osmotic imbalance across the lysosomal membrane, resulting in recruitment of LC3-II onto the membrane of the endolysosome or lysosome.
Cathepsins are lysosomal enzymes that play a critical role in protein degradation, including during autophagy. CTSD is activated under the acidic conditions of the lysosome [Citation77], and can be translocated from the lysosome to the cytoplasm, where it mediates apoptosis following pro-apoptotic stimuli including interferon-gamma [Citation78], Fas/APO-1 [Citation78], TNF-alpha [Citation78], and staurosporine [Citation79]. LAMP3 expression could mediate an increase in cytoplasmic CTSD by decreasing LAMP1 protein. In addition, other cathepsins such as CTSB were also released into the cytoplasm in LAMP3-OE cells.
DQ-BSA and galectin puncta formation assays are useful in evaluating the state of the lysosome. DQ-BSA, a reporter for protease activity in the lysosome, is bovine serum albumin labeled with fluorescent dye and it is self-quenched during steady state. DQ-BSA will accumulate in the lysosome and following proteolysis is dequenched, releasing fluorescent fragments [Citation80,Citation81]. Galectins, a marker of pore formation in membranes, are soluble carbohydrate-binding lectins found in the cytoplasm and nucleus that are recruited to the site of endo-lysosomal leakage [Citation82]. LAMP3 OE induced a decrease in DQ-BSA fluorescence and an increase in the percent of LGALS3-puncta positive cells compared with control cells () suggesting that LAMP3 induced the decrease in lysosomal function by the decrease in lysosomal integrity. These results suggest that LAMP3 inhibited the downstream processes of autophagy by inhibiting the function of lysosomal enzymes, which is important in the progression of autophagy, and that LAMP3 induced the release of lysosomal enzyme from the lysosome to the cytoplasm.
BID [Citation65] and caspase 8 [Citation83], proteins in the apoptosis pathway, are known to be activated by CTSD. Cleaved BID leads to released CYCS/cytochrome c from mitochondria resulting in cell death [Citation83–86]. LAMP3-induced activation of BID and CASP3 by CTSD and the LAMP3-induced apoptosis was inhibited by LAMP1 OE and CTSD inhibitor pepA as well as the pan-caspase inhibitor Z-VAD and CASP3-specific inhibitor Z-DEVD (). These results suggest that LAMP3-induced apoptosis via CTSD-BID-CASP3 pathway as a result of the release of CTSD into the cytoplasm following lysosomal membrane instability induced by a decrease of LAMP1 protein. Moreover, LAMP3 induced the release of CTSB, which is associated with cell death following the activation of CASP1 and could be blocked with the CTSB inhibitor, E64D, as well as the CASP1-specific inhibitor, Y-VAD (). Released cathepsins could serve as a mechanism for further loss of LAMP1 as cathepsins OE in the cytoplasm is associated with increased susceptibility to lysosomal membrane permeabilization [Citation87].
Recent papers indicate that LAMP3 has a role in autophagy and LAMP3 knockdown cells may decrease autophagic flux [Citation88,Citation89]. A key observation in our study was that LAMP3 OE inhibited autophagic flux. The mechanism of inhibition involved the breakdown of the lysosome and release of lysosomal enzymes into the cytoplasm impairing the downstream steps of autophagy. Cell death by lysosomal membrane permeabilization and release of cathepsins into the cytoplasm has been previously reported [Citation57]. However, the mechanism of action is not well understood. Experimental agents that induce cell death via lysosomal membrane permeabilization are typically compounds that sensitize cancer cells to die rather than the induction of endogenous pathways [Citation57,Citation90]. Furthermore, lysosomal membrane permeabilization can induce cell death via several mechanisms including both caspase-dependent and independent, and can activate the inflammasome releasing cytokines [Citation86,Citation91,Citation92]. A common characteristic of lysosomal membrane permeabilization-associated cell death is the sensitivity to cathepsin inhibitors such as pepA [Citation91]. Our observation of pepA-sensitive activated CASP3 supported a death pathway typically associated with lysosome-dependent cell death. Indeed, cytoplasmic cathepsins are reported to cleave BID and the translocation of the proapoptotic proteins BAX and BAK1, which in turn induce mitochondrial membrane permeabilization and caspase-dependent apoptosis [Citation93].
A central question remaining is the underlying cause of the lysosomal membrane permeabilization initiated by LAMP3. Although LAMP1 expression levels are decreased, the protein could be detected suggesting that LAMP3 containing vesicles segregated from LAMP1 containing ones. This observation ruled out a complete rupture of the lysosome and indicates a more specific release of enzymes into the cytoplasm of LAMP3 containing vesicles. Lysosomal membrane destabilization can occur from several factors such as elevated hydrogen peroxide in the cell that reacts with the iron, which accumulates in the lysosome, creating free radicals and lysosomal membrane instability [Citation94]. Additional factors that can affect lysosomal membrane stability include lipid composition, ER stress, or activation of calpains. The mechanism associated with LAMP3-induced lysosomal membrane permeabilization may involve its interaction with other proteins such as HSPA. HSPA affects lysosomal membrane stability by promoting the binding between the lipid bis monoacylglycerol phosphate and acid sphingomyelinase. Acid sphingomyelinase, which mediates the degradation of sphingomyelinase to ceramide and decreases in sphingomyelin levels reduce lysosomal membrane stability [Citation95]. However, in LAMP3-OE HSG and A253 cells HSPA was not inhibited (Figure S3E,F), and LAMP3 OE do not induce ER stress [Citation42]. These results support that LAMP3-induced apoptosis via lysosomal membrane permeabilization was likely dependent on changes in LAMP1 protein levels. This hypothesis was supported by our data demonstrating rescue from lysosomal membrane damage and cell death via LAMP3 OE by expression of LAMP1 ().
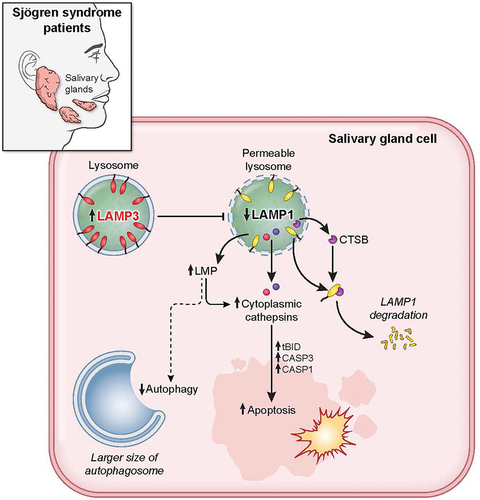
In summary, LAMP3 OE in salivary gland epithelial cells contributed to lysosomal membrane permeabilization mediated apoptosis in SS. At a mechanistic level, LAMP3 expression contributed to a blockade of autophagic flux and lysosomal membrane destabilizing, resulting in cell death by apoptosis following the relocalization of cathepsins into the cytoplasm (). Clarifying the cross talk between LAMP3 expressing cells in the salivary gland and the immune cells as well as surrounding epithelia is required to better understand the underlying pathogenesis in SS. These results defined a causative role for LAMP3 in the pathogenesis of SS and provided support for targeting therapies that block the effect of LAMP3 to prevent cellular apoptosis and inflammation in pSS and the development of autoimmunity.
Figure 8. LAMP3 inhibited autophagy and induced apoptosis via lysosomal membrane permeabilization. LAMP3 OE in minor salivary gland of SS patient leads to lysosomal membrane permeabilization via decreasing LAMP1 protein integrity and its degradation by CTSB, resulting in inhibited autophagy, and cathepsins relocalization. Relocalized cathepsins (CTSB and CTSD) induces the increase in cell death by caspase-dependent apoptosis.
Materials and methods
Patient tissues and clinical information
Formalin-fixed, paraffin embedded salivary gland biopsies from patients being investigated for SS according to the 2002 American-European Consensus Group classification criteria [Citation44] were procured after informed consent as described previously in [Citation42].
Gene expression patterns associated with SS UPR dysfunction
Raw data from Yin et al. [Citation69] were normalized using 75th percentile shift normalization and analyzed using custom Python scripts in the Jupyter notebook suite using the SKLEARN, Matplotlib, Seaborn, Numpy and Pandas software libraries. Briefly described, the normalized gene expression profiles were a subset based on the UPR gene pathway provided by the Jax Labs Gene Ontology browser (GO:0030968), transformed into a Pandas dataframe and scaled by subtracting the minimum value present in each gene expression profile and then dividing by the maximum to allow for the effective clustering of genes present at variable expression levels. The resulting data were clustered and visualized using the Seaborn clustermap function. Individual genes exhibiting elevated expression in the SS class were transformed using the pandas melt function for visualization as individual data points using the Seaborn swarm plot function.
Reagents and transfections
Mycoplasma-free cells were cultured and transfected as described previously in [Citation42].
A variety of mammalian expression vectors were used including pME18S-empty, pME18S-LAMP3 [Citation42,Citation96] and pAAV2-nls-EGFP (AAV2-GFP) [Citation97,Citation98] as described previously. pZW005-RFLC3 plasmid was kindly provided by Dr. Joseph Hill (UT Southwestern University). pCMV6-XL5 (PCMV6XL5), pCMV6-LAMP1 (SC116652), pLenti-C-Myc-DDK-P2A-Puro (PS100092), LAMP3-encoding plasmid (RC205344L3), pLenti-C-mGFP-P2A-Puro (PS100093) and LAMP1-mGFP encoding plasmid (RC219208L4) were purchased from OriGene. These plasmids were purified using Endofree plasmid maxi kit (QIAGEN, 12362) and transfected using Lipofectamine 3000 (Thermo Fisher Scientific, L3000075). HSG control cells for HSG-LAMP3 cells (HSG-control cells), HSG-LAMP3 cells, A253 control cells for A253-LAMP3 cells (A253-control cells), and A235-LAMP3 cells were stably propagated under 1 μg/ml puromycin selection (Thermo Fisher Scientific, A1113803).
For GFP degradation experiments, 5 × 105 HSG-control, HSG-LAMP3, A253-control, and A253-LAMP3 cells were transfected with 1.5 μg pZW005-PFLC3 plasmid using Lipofectamine 3000. These cells were used 72 h after transfection.
For RFP-GFP-LC3 experiments, 4.5 × 106 HSG cells were transfected with 9 μg pZW005-RFLC3 plasmid using Lipofectamine 3000. Twenty-four hours after transfection, 5 × 105 pZW005-RFLC3 plasmid transfected HSG cells were transfected with a total amount of 1.5 μg pME18S-empty and pME18S-LAMP3 plasmid using Lipofectamine 3000. These cells were used 48 h after final transfection.
Subcellular fractionation
Subcellular fractionation was performed according to modified protocol of Johansson et al. [Citation79] and Kreuzaler et al. [Citation99] to collect cytoplasmic and membrane-bound organelle fraction. Briefly, cells were lysed by incubation for 10 min in subcellular fractionation buffer (250 mM sucrose [Sigma-Aldrich, S9378], 20 mM HEPES, pH 7.3 [Quality Biological, 118-089-721], 10 mM KCl [Avantor, MK6858-04], 1.5 mM MgCl2 [Quality Biological, 340-034-721], 1 mM EDTA [Corning, 46-034-CI], 1 mM EGTA [Sigma-Aldrich, E3889]) containing 25 μg/ml digitonin (Sigma-Aldrich, D141), protease inhibitor and phosphatase inhibitor (Thermo Fisher Scientific, 87786) at 4°C with gentle shacking. Lysate was centrifuged at 700 g for 12 min at 4°C to eliminate debris and nuclei, and then the supernatant was centrifuged at 10,000 g for 35 min at 4°C. Supernatant after the final centrifugation step was used as cytosolic protein fraction. The pellet was washed in subcellular fractionation buffer and used as the membrane-bound organelle fraction, which included lysosomal proteins.
CTSD activity assay
Cytoplasmic fractions were collected in a subcellular fractionation buffer containing 25 μg/ml digitonin excluding protease inhibitor and phosphatase inhibitor according to the method as described above. Cytoplasmic CTSD activity was measured using a reaction buffer at pH 7.4 (PBS; Thermo Fisher Scientific, 10010023) or reaction buffer provided in the CTSD activity assay kit (Abcam, Ab65302) according to the manufactures’ protocol. The results were normalized to the total protein amount in each cytoplasmic fraction.
Cycloheximide chase
To measure LAMP1 protein degradation, 48 h posttransfection, culture medium was replaced by medium containing 50 μg/ml CHX (Sigma-Aldrich, C7698) following pretreatment with 10 μM pepA (Enzo Life Sciences, ALX-260-085), 10 μM E64D (Enzo Life Sciences, BML-PI107), 10 μM MG132 (Enzo Life Sciences, BML-PI102), 1 μM or 10 μM VR23 (Selleck Chemicals, S7933), or 10 nM or 100 nM IXA (Selleck Chemicals, S2181) for 1 h and after 0, 2, 4, and 8 h of incubation, cells were washed in ice-cold PBS, lysed by incubation in RIPA buffer (Thermo Fisher Scientific, PI89900) supplemented with protease inhibitor and phosphatase inhibitor for 30 min on ice, and cleared by centrifugation at 17,000 g for 25 min at 4°C. Supernatants were heated for 10 min at 97°C in LDS sample buffer (Thermo Fisher Scientific, NP0007). SDS-PAGE and blotting were performed as described below. Each protein expression was normalized to TUBA, and then the ratio of each protein was plotted.
Western blotting
Western blotting was performed as described previously in [Citation42]. Antibodies were summarized in . Briefly, cultured cells were washed in ice-cold PBS, lysed by incubation for 30 min on ice in RIPA buffer supplemented with protease inhibitor, phosphatase inhibitor, and 25 μM CQ (Sigma-Aldrich, C6628), and then cleared by centrifugation at 17,000 g for 25 min at 4°C. The supernatants were heated for 10 min at 97°C in LDS sample buffer, resolved by SDS-PAGE, and electrophoretically transferred to PVDF membranes (Invitrolon PVDF; Thermo Fisher Scientific, LC2005). After blocking with a blocking buffer (5% nonfat dried milk or bovine serum albumin [BSA; Sigma-Aldrich, A2153] in wash buffer [0.1% Tween-20 {Thermo Fisher Scientific, 003005} in TBS {Quality Biological, 351-086-151}]), the membranes were incubated overnight with primary antibodies. After washing three times with a wash buffer (0.1% Tween-20 in TBS), the membranes were reacted with secondary antibodies for 1 h at room temperature. Finally, the signal was visualized using a Super Signal West Pico Chemiluminescent Substrate (Thermo Fisher Scientific, 34080) or Super Signal West Pico PLUS Chemiluminescent Substrate (Thermo Fisher Scientific, 34580) according to the manufacturer’s protocol.
Table 1. Summary of antibodies used for western blotting.
qRT-PCR analysis
cDNA synthesis and quantitative real-time reverse transcriptase polymerase chain reaction (qRT-PCR) was performed as described in [Citation42] to determine the expression levels of CTSD, TRIM21, BID, and ACTB. Following TaqMan probes were used: ACTB/β-actin, Hs01060665_g1; CTSD/cathepsin D, Hs00157205_m1; BID, Hs00609632_m1; LAMP1, Hs00174766_m1 (Thermo Fisher Scientific, Waltham, MA, USA).
Confocal microscopy
Cells were cultured in 8-well Lab-Tek II chamber slides (Thermo Fisher Scientific, 154534PK), washed, fixed, and permeabilized as described previously [Citation42]. Cells for detection of ATP6V1A were fixed with −20°C methanol (Fisher Scientific, A-452-4) for 10 min after washing three times in PBS, and then washed twice in PBS. Cells were incubated overnight with primary antibodies: anti-LAMP3 polyclonal antibody (pAb; Proteintech, Inc., 12632-1-AP) direct labeled with Dylight 488 (Innova Biosciences Ltd., 322-0030), anti-ATP6V1A monoclonal antibody (mAb; Abcam, ab199326) direct labeled with Alexa Fluor 647 antibody (Novus Biologicals, 336-0030) or anti-LAMP1 mAb (Developmental Studies Hybridoma Bank, H4A3), and anti-CTSD pAb (Abcam, ab826) direct labeled with Dylight 594 (Innova Biosciences Ltd., 324-0030) or anti-LC3B pAb (Abgent, AP1802a) direct labeled with Dylight 594. After washing with PBS, cells were incubated with anti-mouse Alexa Fluor 647 antibody (Jackson ImmunoResearch Laboratories, Inc., 715-605-150) at room temperature. Cells were then washed five times with PBS and then mounted. Areas with multiple LAMP3 transfected cells were imaged using an Olympus confocal microscope with silicone immersion 100× objective with 60 stacked sections at a z-dimension of 0.25 µm per section. Image analysis was performed using ICY (v1.9.8) applications.
Galectin puncta assay
Cells were replated on a well in 8-well Lab-Tek II chamber slide 24 h posttransfection. Twenty-four hours after replating, cells were fixed by treatment with 4% paraformaldehyde (Electron Microscopy Sciences, 15710) with PBS for 15 min at 22°C, and then washed twice with PBS for 5 min each. Fixed cells were permeabilized by treatment with 0.1% Triton X-100 (Sigma-Aldrich, 93443) in PBS for 10 min at 22°C, and then incubated with 2% BSA in PBS for 30 min at 22°C. After blocking, cells were incubated with the mixture of 10 μg/ml rabbit anti-LAMP3 pAb and 10 μg/ml mouse anti-LGALS3/galectin 3 mAb (Abcam, ab2785) in 2% BSA in PBS at 4°C overnight. Cells were washed three times in PBS for 10 min each, and then incubated with the mixture of 10 μg/ml Alexa Fluor 647 anti-mouse IgG and 10 μg/ml Alexa Fluor 488 anti-rabbit IgG (Jackson ImmunoResearch Laboratories, Inc., 711-546-152) in 2% BSA in PBS for 1 h at 22°C. After incubation, cells were washed three times in PBS for 10 min each, and then mounted with mounting media with DAPI (Abcam, ab104139). Stained cells were analyzed by using Nikon fluorescent microscope.
Flow cytometry analysis and apoptosis assay
Acidic organelles in control and LAMP3-OE cells of HSG and A253 cells were analyzed using LysoTracker DND-99 (Thermo Fisher Scientific, L7528) according to the manufactures’ instruction. Stained cells detected using the BD Accuri (BD Biosciences, San Jose, CA, USA) and analyzed using BD CSampler software.
Cleaved CASP3 activity was measured using active/cleaved CASP3 Assay Kit (Fluorometric; Novus Biologicals, NBP2-54869) according to the manufacturer’s instruction. Stained cells were detected using the BD Accuri and analyzed using FlowJo (BD Biosciences, San Jose, CA, USA).
The apoptosis assay was performed as described previously in [Citation42]. To study the role of cathepsins and caspases on LAMP3-induced apoptosis, cells were incubated with or without 20 μM Z-VAD-FMK (Z-VAD; Enzo Life Sciences, ALX-260-020), 5 μM pepA, 2.5 μM E64D, 10 μM Z-DEVD-fmk (Z-DEVD; Sigma-Aldrich, 264155), or 50 μM Ac-YVAD-cmk (Y-VAD; Sigma-Aldrich, SML0429) for 14 h preflow cytometry analysis. Stained cells were detected using the BD Accuri and analyzed using FlowJo.
Long-lived protein degradation assay
Long-lived protein degradation assay was performed according to the protocol of Jigang et al. [Citation51] to analyze autophagic protein degradation. Briefly, 2.4 × 106 HSG-control or HSG-LAMP3 cells were plated in three wells in 6-well plates (8 × 105 cells/well). Twenty-four hours postplating, cells were washed twice in warmed PBS, and then incubated with 2 ml of l-methionine-free culture DMEM (l-methionine-, l-cystatin-, pyruvate-, and glutamine-free DMEM [Thermo Fisher Scientific, 21013024]) supplemented with 2% l-cysteine (Sigma-Aldrich, 57579-5ML-F), 1% pyruvate (Thermo Fisher Scientific, 11360070), 2% l-glutamine (Thermo Fisher Scientific, A2916801) with 10% dialyzed FBS (Thermo Fisher Scientific, A3382001) for 30 min at 37°C, 5% CO2. After incubation, the culture medium was replaced with 2 ml of l-methionine-free culture DMEM with 10% dialyzed FBS and 50 μM l-azidohomoalanine (AHA; Thermo Fisher Scientific, C10102). After incubation for 18 h at 37°C, 5% CO2, culture medium was replaced with a regular DMEM with 10% FBS containing 10 × l-methionine (2 mM final concentration; Sigma-Aldrich, M9625) for 2 h at 37°C, 5% CO2. Cells were washed once in warmed-PBS and incubated with regular DMEM with 10% FBS containing 10 × l-methionine (2 mM final concentration) for 3 h at 37°C, 5% CO2. Three hours post-incubation, cells were collected to a 2.0 ml low protein-binding tube (Thermo Fisher Scientific, 88379) by trypsin treatment, washed once in cold-PBS, and then fixed by 1 ml of 4% paraformaldehyde in PBS and incubate for 15 min at 22°C. Cells were washed each once in 1 ml 3% BSA (Sigma-Aldrich, A7638) in PBS and 1 ml cold-PBS, and then incubated with 0.5 ml of 0.5% Triton X-100 in PBS for 20 min at 22°C. After incubation, cells were washed each once in cold-PBS and 3% BSA in PBS. After washing once more in cold-PBS, cells incubated in the click reaction buffer (50 μM Alexa Fluor 647 Alkyne, Triethylammonium Salt [Thermo Fisher Scientific, A10278], 1 mM Tris[2-carboxyethyl]phosphine hydrochloride [TCEP; Sigma-Aldrich, C4706], 100 μM Tris[{1-benzyl-1H-1,2,3-triazol-4-yl}methyl]amine [TBTA; Sigma-Aldrich, 678937] and 1 mM copper(II) sulfate [Sigma-Aldrich, 451657] in PBS) in the dark for 2 h at 24°C with gently shaking at 700 rpm. After washing once in 3% BSA in PBS and twice in cold-PBS, stained cells were detected using the BD Accuri and analyzed using FlowJo.
Analysis of lysosomal degradation capacity
Forty-eight hours posttransfection, A253 control and LAMP3-OE cells were collected and resuspended with 10 μg/mL DQ Green BSA (Thermo Fisher Scientific, D12050) in Mccoy’s 5A (Thermo Fisher Scientific, 16600082) supplemented with 10% FBS. After incubation for 2 h at 37°C, 5% CO2, cells were washed three times in cold-DPBS (+Ca2+ and Mg2+). Stained cells were detected using the BD Accuri and analyzed using FlowJo.
Statistical analysis
Statistical analysis of microarray studies was performed as described previously in [Citation69]. Student’s t-test and ANOVA tests were used for analysis of studies where appropriate. These analyses were carried out with JMP 13.2.0, the Data analysis tool in Excel 2016 and GraphPad Prism (GraphPad Software, San Diego, CA, USA). A p value of less than 0.05 was regarded as statistically significant. All statistical tests were two-sided.
Human subjects research declaration
All clinical investigations were conducted in accordance with the Declaration of Helsinki principles. Written informed consent to IRB-approved protocols (as described above) were obtained from all participants prior to inclusion in the studies described herein. All human studies were approved by the appropriate institutional review board.
Supplemental Material
Download Zip (12.6 MB)Acknowledgments
The authors would like to thank the members of the AAV biology section NIDCR/NIH for assistance with this work and comments and suggestions from Drs. Peter Burbelo and Alan Baer. This work was supported by the Division of Intramural Research, NIDCR/NIH (1ZIADE000695, JAC); JSPS Research Fellowship for Japanese Biomedical and Behavioral Researchers at NIH (71713, TT).
Disclosure statement
No potential conflict of interest was reported by the author(s).
Supplementary material
Supplemental data for this article can be accessed here
Additional information
Funding
Related Research Data
References
- Nocturne G, Mariette X. Advances in understanding the pathogenesis of primary Sjögren’s syndrome. Nat Rev Rheumatol. 2013;9(9):544–556.
- Mariette X, Criswell LA. Primary Sjögren’s syndrome. N Engl J Med. 2018;379(1):97.
- Yildiz Ç, Karakuş S, Bozoklu Akkar Ö, et al. Primary Sjögren’s syndrome adversely affects the female sexual function assessed by the female sexual function index: a case-control study. Arch Rheumatol. 2017;32(2):123–128.
- Leehan KM, Pezant NP, Rasmussen A, et al. Minor salivary gland fibrosis in Sjögren’s syndrome is elevated, associated with focus score and not solely a consequence of aging. Clin Exp Rheumatol. 2018;36 Suppl 112(3):80–88.
- Ramos-Casals M, Font J. Primary Sjögren’s syndrome: current and emergent aetiopathogenic concepts. Rheumatology (Oxford). 2005;44(11):1354–1367.
- Manganelli P, Fietta P. Apoptosis and Sjögren syndrome. Semin Arthritis Rheum. 2003;33(1):49–65.
- Alessandri C, Ciccia F, Priori R, et al. CD4 T lymphocyte autophagy is upregulated in the salivary glands of primary Sjögren’s syndrome patients and correlates with focus score and disease activity. Arthritis Res Ther. 2017;19(1):178.
- Byun YS, Lee HJ, Shin S, et al. Elevation of autophagy markers in Sjögren syndrome dry eye. Sci Rep. 2017;7(1):17280.
- Song S, Tan J, Miao Y, et al. Crosstalk of autophagy and apoptosis: involvement of the dual role of autophagy under ER stress. J Cell Physiol. 2017;232(11):2977–2984.
- Li YY, Feun LG, Thongkum A, et al. Autophagic mechanism in anti-cancer immunity: its pros and cons for cancer therapy. Int J Mol Sci. 2017;18(6):1297.
- Kondo Y, Kanzawa T, Sawaya R, et al. The role of autophagy in cancer development and response to therapy. Nat Rev Cancer. 2005;5(9):726–734.
- Yin H, Wu H, Chen Y, et al. The therapeutic and pathogenic role of autophagy in autoimmune diseases. Front Immunol. 2018;9:1512.
- Webb JL, Ravikumar B, Atkins J, et al. Alpha-Synuclein is degraded by both autophagy and the proteasome. J Biol Chem. 2003;278(27):25009–25013.
- Vogiatzi T, Xilouri M, Vekrellis K, et al. Wild type alpha-synuclein is degraded by chaperone-mediated autophagy and macroautophagy in neuronal cells. J Biol Chem. 2008;283(35):23542–23556.
- Cho MH, Cho K, Kang HJ, et al. Autophagy in microglia degrades extracellular β-amyloid fibrils and regulates the NLRP3 inflammasome. Autophagy. 2014;10(10):1761–1775.
- Du F, Yu Q, Yan S, et al. PINK1 signalling rescues amyloid pathology and mitochondrial dysfunction in Alzheimer’s disease. Brain. 2017;140(12):3233–3251.
- Qi YY, Zhou XJ, Zhang H. Autophagy and immunological aberrations in systemic lupus erythematosus. Eur J Immunol. 2019;49(4):523–533.
- Wu ZZ, Zhang JJ, Gao CC, et al. Expression of autophagy related genes mTOR, Becline-1, LC3 and p62 in the peripheral blood mononuclear cells of systemic lupus erythematosus. Am J Clin Exp Immunol. 2017;6(1):1–8.
- Martinez J, Cunha LD, Park S, et al. Noncanonical autophagy inhibits the autoinflammatory, lupus-like response to dying cells. Nature. 2016;533(7601):115–119.
- Galluzzi L, Vitale I, Abrams JM, et al. Molecular definitions of cell death subroutines: recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ. 2012;19(1):107–120.
- Noguchi M, Hirata N, Tanaka T, et al. Autophagy as a modulator of cell death machinery. Cell Death Dis. 2020;11(7):517.
- Fehrenbacher N, Bastholm L, Kirkegaard-Sørensen T, et al. Sensitization to the lysosomal cell death pathway by oncogene-induced down-regulation of lysosome-associated membrane proteins 1 and 2. Cancer Res. 2008;68(16):6623–6633.
- Salaun B, De Saint-vis B, Pacheco N, et al. CD208/dendritic cell-lysosomal associated membrane protein is a marker of normal and transformed type II pneumocytes. Am J Pathol. 2004;164(3):861–871.
- Berger C, Hoffmann K, Vasquez JG, et al. Rapid generation of maturationally synchronized human dendritic cells: contribution to the clinical efficacy of extracorporeal photochemotherapy. Blood. 2010;116(23):4838–4847.
- Burton TD, Fedele AO, Xie J, et al. The gene for the lysosomal protein LAMP3 is a direct target of the transcription factor ATF4. J Biol Chem. 2020;295(21):7418–7430.
- Mujcic H, Rzymski T, Rouschop KM, et al. Hypoxic activation of the unfolded protein response (UPR) induces expression of the metastasis-associated gene LAMP3. Radiother Oncol. 2009;92(3):450–459.
- Nagelkerke A, Bussink J, van der Kogel AJ, et al. The PERK/ATF4/LAMP3-arm of the unfolded protein response affects radioresistance by interfering with the DNA damage response. Radiother Oncol. 2013;108(3):415–421.
- Lee EJ, Park KS, Jeon IS, et al. LAMP-3 (Lysosome-Associated Membrane Protein 3) promotes the intracellular proliferation of Salmonella typhimurium. Mol Cells. 2016;39(7):566–572.
- Lanford RE, Guerra B, Lee H, et al. Genomic response to interferon-alpha in chimpanzees: implications of rapid downregulation for hepatitis C kinetics. Hepatology. 2006;43(5):961–972.
- Irudayam JI, Contreras D, Spurka L, et al. Characterization of type I interferon pathway during hepatic differentiation of human pluripotent stem cells and hepatitis C virus infection. Stem Cell Res. 2015;15(2):354–364.
- Nakamura H, Kawakami A. What is the evidence for Sjögren’s syndrome being triggered by viral infection? Subplot: infections that cause clinical features of Sjögren’s syndrome. Curr Opin Rheumatol. 2016;28(4):390–397.
- Nakamura H, Shimizu T, Kawakami A. Role of viral infections in the pathogenesis of Sjögren’s syndrome: different characteristics of Epstein-Barr virus and HTLV-1. J Clin Med. 2020;9(5):1459.
- Kompoliti A, Gage B, Sharma L, et al. Human T-cell lymphotropic virus type 1-associated myelopathy, Sjögren syndrome, and lymphocytic pneumonitis. Arch Neurol. 1996;53(9):940–942.
- Ohyama Y, Nakamura S, Hara H, et al. Accumulation of human T lymphotropic virus type I-infected T cells in the salivary glands of patients with human T lymphotropic virus type I-associated Sjögren’s syndrome. Arthritis Rheum. 1998;41(11):1972–1978.
- Toussirot E, Le Huédé G, Mougin C, et al. Presence of hepatitis C virus RNA in the salivary glands of patients with Sjögren’s syndrome and hepatitis C virus infection. J Rheumatol. 2002;29(11):2382–2385.
- Triantafyllopoulou A, Moutsopoulos H. Persistent viral infection in primary Sjogren’s syndrome: review and perspectives. Clin Rev Allergy Immunol. 2007;32(3):210–214.
- Lee SJ, Lee JS, Shin MG, et al. Detection of HTLV-1 in the labial salivary glands of patients with Sjögren’s syndrome: a distinct clinical subgroup? J Rheumatol. 2012;39(4):809–815.
- Wang Y, Dou H, Liu G, et al. Hepatitis C virus infection and the risk of Sjögren or sicca syndrome: a meta-analysis. Microbiol Immunol. 2014;58(12):675–687.
- Litwin CM, Rourk AR. Anti-ENA antibody profiles in patients with hepatitis C virus infection. J Clin Lab Anal. 2018;32(3):e22279.
- Nakamura H, Fujieda Y, Yasuda S, et al. Remission of nephrotic syndrome after therapy for chronic hepatitis C virus infection in a patient with systemic lupus erythematosus. Ann Intern Med. 2018;169(5):352–353.
- Sanosyan A, Daien C, Nutz A, et al. Discrepancy of serological and molecular patterns of circulating Epstein-Barr virus reactivation in primary Sjögren’s syndrome. Front Immunol. 2019;10:1153.
- Tanaka T, Warner BM, Odani T, et al. LAMP3 induces apoptosis and autoantigen release in Sjögren’s syndrome patients. Sci Rep. 2020;10(1):15169.
- Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell. 2010;140(3):313–326.
- Vitali C, Bombardieri S, Jonsson R, et al. Classification criteria for Sjögren’s syndrome: a revised version of the European criteria proposed by the American-European Consensus Group. Ann Rheum Dis. 2002;61(6):554–558.
- Shiboski CH, Shiboski SC, Seror R, et al. 2016 American college of rheumatology/European league against rheumatism classification criteria for primary Sjogren’s syndrome: a consensus and data-driven methodology involving three international patient cohorts. Arthritis Rheumatol. 2017;69(1):35–45.
- Xia SW, Wang ZM, Sun SM, et al. Endoplasmic reticulum stress and protein degradation in chronic liver disease. Pharmacol Res. 2020;161:105218.
- Li Z, Ji X, Wang D, et al. Autophagic flux is highly active in early mitosis and differentially regulated throughout the cell cycle. Oncotarget. 2016;7(26):39705–39718.
- Klionsky DJ, Abdel-Aziz AK, Abdelfatah S, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (4th edition). Autophagy. 2021;17(1):1–382.
- Shintani T, Klionsky DJ. Cargo proteins facilitate the formation of transport vesicles in the cytoplasm to vacuole targeting pathway. J Biol Chem. 2004;279(29):29889–29894.
- Eng CH, Yu K, Lucas J, et al. Ammonia derived from glutaminolysis is a diffusible regulator of autophagy. Sci Signal. 2010;3(119):ra31.
- Wang J, Zhang J, Lee YM, et al. Nonradioactive quantification of autophagic protein degradation with L-azidohomoalanine labeling. Nat Protoc. 2017;12(2):279–288.
- Lee DE, Bareja A, Bartlett DB, et al. Autophagy as a therapeutic target to enhance aged muscle regeneration. Cells. 2019;8(2):183.
- du Toit A, Hofmeyr JS, Gniadek TJ, et al. Measuring autophagosome flux. Autophagy. 2018;14(6):1060–1071.
- Tsubuki S, Saito Y, Tomioka M, et al. Differential inhibition of calpain and proteasome activities by peptidyl aldehydes of di-leucine and tri-leucine. J Biochem. 1996;119(3):572–576.
- Lee DH, Goldberg AL. Proteasome inhibitors: valuable new tools for cell biologists. Trends Cell Biol. 1998;8(10):397–403.
- Longva KE, Blystad FD, Stang E, et al. Ubiquitination and proteasomal activity is required for transport of the EGF receptor to inner membranes of multivesicular bodies. J Cell Biol. 2002;156(5):843–854.
- Boya P, Kroemer G. Lysosomal membrane permeabilization in cell death. Oncogene. 2008;27(50):6434–6451.
- Song XB, Liu G, Liu F, et al. Autophagy blockade and lysosomal membrane permeabilization contribute to lead-induced nephrotoxicity in primary rat proximal tubular cells. Cell Death Dis. 2017;8(6):e2863.
- Kirkegaard T, Roth AG, Petersen NH, et al. Hsp70 stabilizes lysosomes and reverts Niemann-Pick disease-associated lysosomal pathology. Nature. 2010;463(7280):549–553.
- Nylandsted J, Gyrd-Hansen M, Danielewicz A, et al. Heat shock protein 70 promotes cell survival by inhibiting lysosomal membrane permeabilization. J Exp Med. 2004;200(4):425–435.
- Eriksson I, Wäster P, Öllinger K. Restoration of lysosomal function after damage is accompanied by recycling of lysosomal membrane proteins. Cell Death Dis. 2020;11(5):370.
- Clarke P, Tyler KL. Apoptosis in animal models of virus-induced disease. Nat Rev Microbiol. 2009;7(2):144–155.
- Li C, Dai L, Zhang J, et al. Follistatin-like protein 5 inhibits hepatocellular carcinoma progression by inducing caspase-dependent apoptosis and regulating Bcl-2 family proteins. J Cell Mol Med. 2018;22(12):6190–6201.
- Hentze H, Lin XY, Choi MS, et al. Critical role for cathepsin B in mediating caspase-1-dependent interleukin-18 maturation and caspase-1-independent necrosis triggered by the microbial toxin nigericin. Cell Death Differ. 2003;10(9):956–968.
- Heinrich M, Neumeyer J, Jakob M, et al. Cathepsin D links TNF-induced acid sphingomyelinase to Bid-mediated caspase-9 and −3 activation. Cell Death Differ. 2004;11(5):550–563.
- Clerc P, Jeanjean P, Hallali N, et al. Targeted Magnetic Intra-Lysosomal Hyperthermia produces lysosomal reactive oxygen species and causes Caspase-1 dependent cell death. J Control Release. 2018;270:120–134.
- Jia C, Zhang J, Chen H, et al. Endothelial cell pyroptosis plays an important role in Kawasaki disease via HMGB1/RAGE/cathespin B signaling pathway and NLRP3 inflammasome activation. Cell Death Dis. 2019;10(10):778.
- Greenwell-Wild T, Moutsopoulos NM, Gliozzi M, et al. Chitinases in the salivary glands and circulation of patients with Sjögren’s syndrome: macrophage harbingers of disease severity. Arthritis Rheum. 2011;63(10):3103–3115.
- Yin H, Cabrera-Perez J, Lai Z, et al. Association of bone morphogenetic protein 6 with exocrine gland dysfunction in patients with Sjögren’s syndrome and in mice. Arthritis Rheum. 2013;65(12):3228–3238.
- Tsuboi H, Nakai Y, Iizuka M, et al. DNA microarray analysis of labial salivary glands in IgG4-related disease: comparison with Sjögren’s syndrome. Arthritis Rheumatol. 2014;66(10):2892–2899.
- Nakamura H, Tanaka T, Pranzatelli T, et al. Lysosome-associated membrane protein 3 misexpression in salivary glands induces a Sjögren’s syndrome-like phenotype in mice. Ann Rheum Dis. 2021;80:1031–1039.
- Lumkwana D, Du Toit A, Kinnear C, et al. Autophagic flux control in neurodegeneration: progress and precision targeting-Where do we stand? Prog Neurobiol. 2017;153:64–85.
- Minakaki G, Menges S, Kittel A, et al. Autophagy inhibition promotes SNCA/alpha-synuclein release and transfer via extracellular vesicles with a hybrid autophagosome-exosome-like phenotype. Autophagy. 2018;14(1):98–119.
- de Duve C, De Barsy T, Poole B, et al. Commentary. Lysosomotropic agents. Biochem Pharmacol. 1974;23(18):2495–2531.
- Florey O, Gammoh N, Kim SE, et al. V-ATPase and osmotic imbalances activate endolysosomal LC3 lipidation. Autophagy. 2015;11(1):88–99.
- Repnik U, Hafner Česen M, Turk B. Lysosomal membrane permeabilization in cell death: concepts and challenges. Mitochondrion. 2014;19(Pt):A:49–57.
- Zaidi N, Maurer A, Nieke S, et al. Cathepsin D: a cellular roadmap. Biochem Biophys Res Commun. 2008;376(1):5–9.
- Deiss LP, Galinka H, Berissi H, et al. Cathepsin D protease mediates programmed cell death induced by interferon-gamma, Fas/APO-1 and TNF-alpha. EMBO J. 1996;15(15):3861–3870.
- Johansson AC, Steen H, Ollinger K, et al. Cathepsin D mediates cytochrome c release and caspase activation in human fibroblast apoptosis induced by staurosporine. Cell Death Differ. 2003;10(11):1253–1259.
- Klionsky DJ, Abeliovich H, Agostinis P, et al. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy. 2008;4(2):151–175.
- Frost LS, Dhingra A, Reyes-Reveles J, et al. The use of DQ-BSA to monitor the turnover of autophagy-associated cargo. Methods Enzymol. 2017;587:43–54.
- Aits S, Kricker J, Liu B, et al. Sensitive detection of lysosomal membrane permeabilization by lysosomal galectin puncta assay. Autophagy. 2015;11(8):1408–1424.
- Conus S, Pop C, Snipas SJ, et al. Cathepsin D primes caspase-8 activation by multiple intra-chain proteolysis. J Biol Chem. 2012;287(25):21142–21151.
- Stoka V, Turk B, Schendel SL, et al. Lysosomal protease pathways to apoptosis. Cleavage of Bid, not pro-caspases, is the most likely route. J Biol Chem. 2001;276(5):3149–3157.
- Reiners JJ Jr, Caruso JA, Mathieu P, et al. Release of cytochrome c and activation of pro-caspase-9 following lysosomal photodamage involves Bid cleavage. Cell Death Differ. 2002;9(9):934–944.
- Conus S, Simon HU. Cathepsins: key modulators of cell death and inflammatory responses. Biochem Pharmacol. 2008;76(11):1374–1382.
- Xu Y, Wang J, Song X, et al. Protective mechanisms of CA074-me (other than cathepsin-B inhibition) against programmed necrosis induced by global cerebral ischemia/reperfusion injury in rats. Brain Res Bull. 2016;120:97–105.
- Nagelkerke A, Sieuwerts AM, Bussink J, et al. LAMP3 is involved in tamoxifen resistance in breast cancer cells through the modulation of autophagy. Endocr Relat Cancer. 2014;21(1):101–112.
- Dominguez-Bautista JA, Klinkenberg M, Brehm N, et al. Loss of lysosome-associated membrane protein 3 (LAMP3) enhances cellular vulnerability against proteasomal inhibition. Eur J Cell Biol. 2015;94(3–4):148–161.
- Gonzalez P, Mader I, Tchoghandjian A, et al. Impairment of lysosomal integrity by B10, a glycosylated derivative of betulinic acid, leads to lysosomal cell death and converts autophagy into a detrimental process. Cell Death Differ. 2012;19(8):1337–1346.
- Serrano-Puebla A, Boya P. Lysosomal membrane permeabilization as a cell death mechanism in cancer cells. Biochem Soc Trans. 2018;46(2):207–215.
- Heid ME, Keyel PA, Kamga C, et al. Mitochondrial reactive oxygen species induces NLRP3-dependent lysosomal damage and inflammasome activation. J Immunol. 2013;191(10):5230–5238.
- Oberle C, Huai J, Reinheckel T, et al. Lysosomal membrane permeabilization and cathepsin release is a Bax/Bak-dependent, amplifying event of apoptosis in fibroblasts and monocytes. Cell Death Differ. 2010;17(7):1167–1178.
- Kurz T, Gustafsson B, Brunk UT. Intralysosomal iron chelation protects against oxidative stress-induced cellular damage. FEBS J. 2006;273(13):3106–3117.
- Petersen NH, Kirkegaard T. HSP70 and lysosomal storage disorders: novel therapeutic opportunities. Biochem Soc Trans. 2010;38(6):1479–1483.
- Laine J, Künstle G, Obata T, et al. The protooncogene TCL1 is an Akt kinase coactivator. Mol Cell. 2000;6(2):395–407.
- Schmidt M, Grot E, Cervenka P, et al. Identification and characterization of novel adeno-associated virus isolates in ATCC virus stocks. J Virol. 2006;80(10):5082–5085.
- Craig AT, Gavrilova O, Dwyer NK, et al. Transduction of rat pancreatic islets with pseudotyped adeno-associated virus vectors. Virol J. 2009;6:61.
- Kreuzaler PA, Staniszewska AD, Li W, et al. Stat3 controls lysosomal-mediated cell death in vivo. Nat Cell Biol. 2011;13(3):303–309.