
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
The polymorphism L412F in TLR3 has been associated with several infectious diseases. However, the mechanism underlying this association is still unexplored. Here, we show that the L412F polymorphism in TLR3 is a marker of severity in COVID-19. This association increases in the sub-cohort of males. Impaired macroautophagy/autophagy and reduced TNF/TNFα production was demonstrated in HEK293 cells transfected with TLR3L412F-encoding plasmid and stimulated with specific agonist poly(I:C). A statistically significant reduced survival at 28 days was shown in L412F COVID-19 patients treated with the autophagy-inhibitor hydroxychloroquine (p = 0.038). An increased frequency of autoimmune disorders such as co-morbidity was found in L412F COVID-19 males with specific class II HLA haplotypes prone to autoantigen presentation. Our analyses indicate that L412F polymorphism makes males at risk of severe COVID-19 and provides a rationale for reinterpreting clinical trials considering autophagy pathways.
Abbreviations: AP: autophagosome; AUC: area under the curve; BafA1: bafilomycin A1; COVID-19: coronavirus disease-2019; HCQ: hydroxychloroquine; RAP: rapamycin; ROC: receiver operating characteristic; SARS-CoV-2: severe acute respiratory syndrome coronavirus 2; TLR: toll like receptor; TNF/TNF-α: tumor necrosis factor
Introduction
In December 2019, a new virus was isolated in Wuhan, China, which was called Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2). SARS-CoV-2 is an enveloped positive-sense RNA virus that caused a new pandemic, which WHO named COVID-19 (coronavirus disease-2019).
To date, many characteristics of SARS-CoV-2 are still unclear and, although its ability to be transmitted from one person to another has been ascertained, uncertainties remain about the exact modes of transmission and pathogenicity. In addition, a high variability of symptoms in infected patients and between different populations has been reported; one of the possible explanations of such variability is the genetic background of the host that may affect immune responses to the virus. Among host genetic factors that might impact on symptoms severity there are genes involved in virus entry and mediators of innate immunity [Citation1,Citation2]. TLRs (toll like receptors) are a class of proteins that play a key role in host innate immunity, causing the production of pro-inflammatory cytokines (TNF, IL1, and IL6) and type I and II Interferons, that are responsible for innate antiviral responses. Among TLR genes, TLR3 encodes an interferon‐inducing dsRNA sensor, whose activation is involved in protection against different RNA viruses [Citation2–4]. Upon viral infection, TLR3 signaling leads to the activation of two factors, NFKB and IRF3 (interferon regulatory factor 3), which play an essential role in the immune response. This results in the production of various cytokines, including TNF (tumor necrosis factor), activating immune responses. However, increased inflammatory responses can make the patient more susceptible to pneumonia and autoimmune diseases. Accordingly, a protective effect against fatal pneumonia has been reported in the absence of TLR3 [Citation5–7]. Among TLR3 variants, the functional L412F polymorphism (rs3775291; c.1234 C > T) is known to decrease TLR3 expression on the cell surface [Citation8]. This polymorphism also leads to poor recognition of SARS-CoV-2 dsRNA, during replication, compared to its wild-type (WT) counterpart [Citation9] and has been recently associated with SARS-CoV-2 susceptibility and mortality [Citation10].
There is evidence that TLR3, as with other TLRs, acts through autophagy in determining susceptibility to infections [Citation11]. The autophagic pathway is essential during infection and for molecular processes such as cell maintenance and homeostasis [Citation12,Citation13]. Indeed, autophagy is one of the major cell defense mechanisms against pathogens [Citation14]. A role for autophagy is reported in different studies on other coronaviruses such as the mouse hepatitis virus/MHV and the transmissible gastroenteritis virus/TGEV [Citation15,Citation16]. A role in SARS-CoV-2 infection has also been described [Citation17–19]. In particular, SARS-CoV-2 can inhibit autophagy resulting in accumulation of autophagosomes and inhibition of viral clearance that, together with immune dysfunction and the activation of numerous inflammatory cytokines, leads to a more severe form of COVID-19 [Citation20–22].
To shed light on the mechanisms underlying the diverse susceptibility to COVID-19, we performed a nested-control study within our GEN-COVID cohort, confirming the role of L412F polymorphism in the TLR3 gene in susceptibility to SARS-CoV-2 and further defining the potential mechanisms by which this effect is exerted.
Results and discussion
Comparing the extreme phenotypes of SARS-CoV-2 infection, severe COVID-19 patients (cases) versus SARS-CoV-2 PCR-positive oligo-asymptomatic subjects (controls), and using LASSO Logistic regression on common bi-allelic polymorphisms from whole-exome sequencing, we identified the L412F polymorphism (rs3775291; c.1234 C > T) in TLR3 as a severity marker (). The grid search curve of the cross-validation score () shows a maximum of the regularization parameter in 10. With this calibration setting, the 10-fold cross-validation provides good performances in terms of accuracy (73%), precision (74%), sensitivity (73%), and specificity (73%) as shown in . The confusion matrix is reported in , whereas the receiver operating characteristic (ROC) curve () provides an area under the curve (AUC) score of 80%.
Figure 1. The histogram of the LASSO logistic regression weights represents the importance of each feature for the classification task, (A) The positive weights reflect a susceptible behavior of the features to the target COVID-19 disease, whereas the negative weights a protective action. (B) Cross-validation ROC-AUC score for the grid of LASSO regularization parameters; the error bar is given by the standard deviation of the score within the 10 folds; the optimal regularization parameter is chosen by selecting the one with highest cross-validation score (red point). (C) Boxplot of accuracy, precision, sensitivity, specificity, and ROC-AUC score for the 10-fold of the cross-validation. The box extends from the Q1 to Q3 quartile, with a line at the median (Q2) and a triangle for the average. (D) Confusion matrix for the aggregation of the logistic regression predictions in the 10 folds of the cross-validation. (E) ROC curve for the 10 folds of the cross-validation. (F) Distribution of carriers of the polymorphism L412F in homozygous or heterozygous states stratified by clinical category.
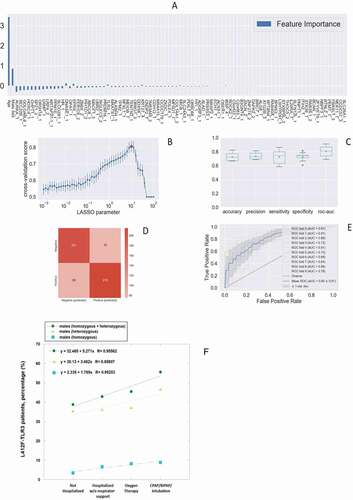
The L412F polymorphism has an overall allele frequency of about 20%, ranging from 30% in European to 0.88% in African (mainly sub Saharan) populations [Citation8]. It is intriguing that a relatively COVID-19-free population such as sub Saharan has a very low frequency (0.88%) of this polymorphism and that Asian (26.97%) and European (30.01%) have a much higher frequency. The variant protein with phenylalanine is under-represented on the cell surface, it is not efficiently secreted into the culture medium when expressed as the soluble ectodomain, and it has reduced capability to activate the expression of TLR3-dependent reporter constructs [Citation8]. In order to confirm the role of the polymorphism, we compared individuals showing severe COVID-19 (cases) and those with no sign of the disease (controls). We subdivided patients into two categories, those having the polymorphism in heterozygous or homozygous state and those homozygous for the WT allele. We found that the prevalence of L412F polymorphism is significantly higher in cases compared to controls (p-value 2.8 × 10−2) (). The global allele frequency of L412F in our cohort (cases and controls) is 29.38%, comparable to the allele frequency of 29.79% reported in the European (non-Finnish) population in the gnomAD database (https://gnomad.broadinstitute.org/). The identified frequencies were in Hardy-Weinberg equilibrium.
Table 1. L412F and COVID-19 outcome (both sexes).
Sex-related differences of TLRs activation following stimulation by viral nucleic acid may be involved in the sex-related variability in response to viral infections [Citation23]. Several rare TLR3 loss of function mutations are known to be linked both to influenza and SARS-CoV-2 virus as well as hyperfunctioning mutations [Citation24,Citation25]. In agreement with these data, when we stratified by gender, the statistically significant difference increased in the sub-cohort of males giving an Odds Ratio of 1.94 (95% confidence interval, 1.23 to 3.06; p = 3.8x10−3), whereas it was lost in the sub-cohort of females (p-value 5.8 × 10−1) ().
Table 2. L412F and COVID-19 outcome (males only).
Table 3. L412F and COVID-19 outcome (females only).
We then investigated the prevalence of patients carrying L412F in heterozygous or homozygous states in all 4 categories of COVID-19 clinical severity, considering only male subjects regardless of age (n = 665). We found that the prevalence of carriers directly increased with the severity of COVID-19, from a clinical condition not-requiring hospitalization to intratracheal intubation ()
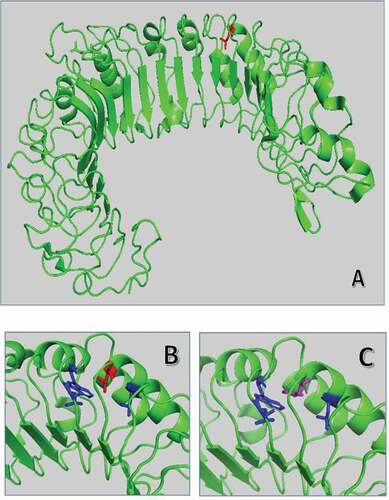
The L412F substitution in TLR3 falls in the ectodomain, in the 14 leucine-rich repeats/LRR domain, a motif of 22 amino acids in length that folds into a horseshoe shape [Citation26]. Proteins containing leucine-rich repeats are involved in a variety of biological processes, including signal transduction, cell adhesion, DNA repair, recombination, transcription, RNA processing, disease resistance, apoptosis, and the immune response [Citation27]. The L412F substitution is expected to have a limited structural impact with minimal rearrangement of near hydrophobic amino acids such as tryptophan 386 (). However, the absence of one of the leucines probably determines a different rearrangement of the motif and consequently of the near glycosylation site Asn414, having an impact on protein-protein interaction and in signal transduction process [Citation28].
Figure 2. Superposition of wild-type and mutated TLR3 protein. (A) TRL3 human protein tridimensional structure of 2Z7X crystal structure. In green cartoon representation of TLR3 protein. (B) and (C) Zoom of the mutated region with Leu412 in red sticks and Phe412 in magenta. The hydrophobic core of Leu377, Leu389, and Trp386 is in blue sticks.
Germline knockout of TLR3 inhibits autophagy and upregulation of TLR3 promotes damage after myocardial infarction mainly because of autophagy rather than inflammatory activation [Citation29]. Interestingly, we could notice a statistically significant (p = 3.8x10−2) reduced survival at 28 days in TLR3_L412F COVID-19 patients treated with hydroxychloroquine (HCQ) (). As this drug is a well-established inhibitor of autophagy, we reasoned that alterations of this important biological process might have a role in the increased severity of the clinical phenotypes of SARS-CoV-2 infection in patients with the TLR3L412F polymorphism. Notably, beside being entirely ineffective at changing the clinical evolution of COVID-19, which led to retraction of the paper reporting the clinical trial [Citation30], HCQ may have been responsible for reportedly increased fatality rates among patients treated with this drug [Citation31,Citation32]. Poly(I:C) stimulation of the TLR3 receptor has already been shown to stimulate autophagy [Citation29]. Therefore, we decided to compare the efficacy of transfected WT and L412F-mutated receptors in inducing autophagy upon poly(I:C) treatment. To monitor autophagy, we used indirect immunofluorescence microscopy to score the formation of punctate intracellular vacuoles stained for LC3B. Moreover, to better appreciate autophagosomal formation, we also used bafilomycin A1 (Baf A1), an inhibitor of lysosomal acidification, to prevent lysosomal degradation of autophagosome-associated LC3B [Citation33]. Baf A1 allows, in fact, the detection of each autophagosome formed in the time-lapse between addition of the drug to cells and harvesting [Citation33]. Importantly, to avoid potentially confounding effects of the endogenous TLR3 receptor in transfected cells, we used TLR3 knockout HEK cells. In these cells, when transfected with a plasmid encoding WT TLR3 (TLR3_WT), we observed a progressively increasing number of autophagosomes (APs) when stimulated with poly(I:C) for different time points in the presence of BAF A1 (compared to BAF A1 alone) (), indicating a stimulation of the synthesis of these vesicles and a positive autophagic flux. Conversely, in HEK cells transfected with TLR3_L412F, the number of AP was reduced by poly(I:C) stimulation in the presence of BAF A1 (), demonstrating a block in AP synthesis and a reduced autophagic flux. Interestingly, in the absence of BAF A1, AP numbers did not increase upon poly(I:C) stimulation, suggesting that, in HEK cells, fast degradation of AP may compensate for a small increase in the synthesis. Indeed, also rapamycin (RAP), a strong stimulus for autophagy [Citation34], induced only a small increase of AP in the absence of BAF A1 in these cells upon transfection of both TLR3_WT and TLR3_L412F, supporting slow rates of AP synthesis in these cells upon stimulation with different stimuli. The fusion process of autophagosomes with hydrolase-containing lysosomes represents the final step in the degradation process along the autophagic route and its evaluation provides important information for flux analysis [Citation33]. Therefore, as a further confirmation of a reduced autophagic flux in HEK cells expressing the TLR3L412F mutant as compared to TLR3_WT-transfected cells, we decided to score fusion of autophagosomes to lysosomes in these cells upon poly(I:C) stimulation, by measuring colocalization rate of LC3B and LAMP-1, through analysis with the Volocity software of immunocytochemistry experiments (Fig. S1).
Figure 3. Analysis of autophagy in TLR3_L412F-expressing cells. (A) 28-day survival study of TLR3- L412F carriers vs not-carriers in the group treated with hydroxychloroquine. N = 156, with 73 carriers of TLR3- L412F. Three carriers and 1 not-carrier died in the first 28 days of treatment. (B) Analysis of autophagy in HEK-KO cells expressing wild type or L412F mutant proteins. HEK-KO cells were transfected for 24 h with plasmids encoding TLR3_WT and TLR3_L412F. Cells were next incubated in full medium (FM) or FM + 400 nM bafilomycin A1 (BAF A1) for 3 h and stimulated for increasing times with 50 μg/ml poly(I:C), as indicated. Cells were next fixed, permeabilized with 100 μg/ml digitonin and stained with anti-LC3B antibodies and revealed with Alexa Fluor 488-conjugated secondary antibodies. Nuclei were stained with DAPI. Where indicated, RAP (500 nM, for 2 h) was used as positive control for induction of autophagy. (C) Same as in B, but the amount of autophagosomes (scored as LC3B-positive dots) per cell was quantified by Volocity software. Measures were obtained by analyzing at least 400 cells/sample from 3 different experiments (n = 3). (D) Analysis of TNF mRNA expression in HEK-KO cells expressing wild type or L412F mutant proteins. HEK-KO cells were transfected for 24 h with plasmids encoding empty vector (CTR), TLR3_WT and TLR3_L412F and next stimulated for increasing times with 50 μg/ml poly(I:C), where indicated. TNF levels were evaluated by Real Time PCR. The gene expression levels were evaluated by the fold change versus TLR2 WT sample using the equation 2−DDCt. Data are presented as the mean ± SEM. Data significance was analyzed using One-way ANOVA test with Holm-Sidak’s correction. Asterisks were attributed for the following significance values: P > 0.05 (ns), P < 0.05 (*) and P < 0.01 (**). (E) Normal human fibroblasts (NDHF) from subjects expressing the TLR3_WT receptor were stimulated with 50 μg/ml poly(I:C) or RAP (1 μ M) for 4 h, in full medium alone or containing 400 nM bafilomycin A1 for 3 h. Cells were next fixed, permeabilized with 100 μg/ml digitonin and stained with anti-LC3B antibodies and revealed with Alexa Fluor 488-conjugated secondary antibodies. Nuclei were stained with DAPI. (F) Same as in E, but the number of autophagosomes (scored as LC3B-positive dots) per cell was evaluated for each sample by Volocity software. (G and H) same as in E-F, but fibroblasts are homozygous for the TLR3_L412F receptor. Statistical analysis was performed using Student’s t test. Means § SEM for each value are shown in the graphs. ns = not significant; ** = p < 0.01; *** = p < 0.001. < 0.001.
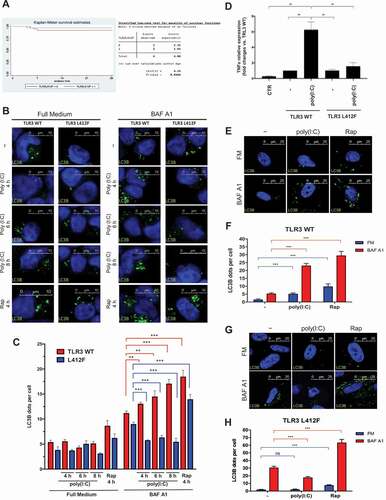
During SARS-CoV-2 infection, adipocytes produce pro-inflammatory cytokines like TNF, IL6 and IL1B/IL-1β which recruit immune cells to the site of infection [Citation35]. Autophagy is stimulated and regulated by these pro-inflammatory cytokines [Citation35]. TNF is a potent immunomodulator and proinflammatory cytokine that has been implicated in the pathogenesis of autoimmune and infectious diseases. It is produced by activated monocytes and macrophages, as well as by many other cell types, including lymphocytes, as a transmembrane protein. Through cell modulation, TNF can activate both cell death and survival mechanisms. TNF induces autophagy through a feedback mechanism, causing further recruitment and activation of lymphocytes and contributing to the excess inflammation typical of SARS-CoV-2 infection [Citation36]. As chloroquine, a powerful inhibitor of autophagy, inhibits production of different cytokines, among which TNF [Citation37], we next decided to test if the inhibitory effect of the L412F mutation on autophagy was also able to mimic the effect of the pharmacological inhibitor of this process in HEK cells. Indeed, while poly(I:C) readily stimulated TNF expression in HEK cells transfected with the TLR3_WT receptor, this effect was completely abolished in TLR3_L412F-transfected cells ().
In order to validate data obtained on transfected HEK cells, we next isolated and cultured skin fibroblasts from healthy donors with different genotypes relative to the TLR3 locus: wild-type (WT/WT) and L412F (L412F/L412F) homozygous. In these primary fibroblasts, immunofluorescence analysis revealed that the number of LC3B-positive vesicles increased upon poly(I:C) stimulation both in the absence and in the presence of BAF A1 (), showing an overall positive autophagic flux in WT cells, while the flux resulted significantly reduced in L412F/L412F fibroblasts (). As a control, RAP stimulation of both WT/WT and L412F/L412F showed a positive autophagic flux () confirming that the mutation specifically affected TLR3-dependent autophagy and not the general autophagic process. Also in these cells, we confirmed a reduced autophagic flux in TLR3_L412F-expressing fibroblasts as compared to TLR3_WT-expressing cells, upon stimulation with poly(I:C), by measurement of the LC3B-LAMP1 colocalization rate (Fig. S2).
Overall, our results therefore suggest that the outcome of clinical trials with HCQ should be reinterpreted in the light of TLR3L412F polymorphism status. Negative effects of the drug in L412F bearing subjects may have masked a possible positive outcome in L412F-free subjects. Importantly, they also support a positive role of autophagy in the anti-viral response of the organism to SARS-CoV-2, as suggested by a recent report by Hayn and colleagues [Citation38], demonstrating that at least 3 viral proteins are able to specifically block autophagic turnover.
TLR3 variant L412F has been associated with a wide range of autoimmune diseases including Addison disease and hypothyroidism [Citation39]. TLR3 rare variants resulting in partial loss of function and occurring together with the common variant L412F, or with another rare variant, have been identified in Addison disease [Citation40]. Persistent viral infections in a background of defective innate immunity lead to overexpression of HLA allotypes prone to present autoantigen. Defects of autophagy have been observed in many infectious and autoimmune diseases. Alteration of autophagic processes causes the onset of autoimmunity due to increased survival and reduced apoptosis of self-reactive lymphocytes [Citation41–43]. HLA has been shown to be implicated in disease severity and clinical outcome of patients with COVID-19 [Citation44]. Accordingly, an increased frequency of autoimmune disorders as co-morbidity was found in our cohort in L412F COVID-19 patients with specific HLA class II haplotypes prone to autoantigen presentation. In particular, we analyzed the DR3-DQ2 haplotype which predisposes to different types of autoimmune diseases [Citation45,Citation46]. The frequency of autoimmune disorders is indeed significantly increased in male patients with HLA DR3/DQ2 haplotype and L412F, especially diabetes (25%) ( and ). These results suggest that the combination of L412F in TLR3 and a specific class II HLA haplotype puts male patients at risk of post-COVID autoimmune exacerbation emphasizing the need for appropriate follow-up.
Table 4. Association between DR3-DQ2 + L412F haplotype and autoimmune disorders in male patients.
Table 5. Male patients with L412F and HLA DR3/DQ2 haplotype.
No association was found between AIRE loss of function variants and COVID-19 outcome, as outlined by the absence of the gene in .
In conclusion, we have identified the second protein-encoding polymorphism that modulates COVID-19 outcome. These results indicate that L412F polymorphism in the TLR3 gene makes males, in whom after puberty testosterone lowers TLR3 expression, at risk of severe COVID-19 in a context of a polygenic model. Moreover, based on impairment of autophagy, these data provide a rationale for reinterpreting clinical trials with HCQ stratifying patients by L412F. Finally, the combination of L412F in TLR3 and specific HLA class II haplotypes may put male patients at risk of post-acute sequelae of SARS-CoV-2 infection pointing to the need for an appropriate follow-up. Our experiments suggest an important role of autophagy downstream of the TLR3 receptor, possibly affecting TNF production and susceptibility to infections, including SARS-CoV-2, pinpointing to IFNs treatment (especially IFN γ) avoiding hydroxiclorochine.
Materials and methods
Patients
We performed a nested case-control study (NCC). We used a cohort of 1319 subjects (cases and controls) from the Italian GEN-COVID Multicenter study, infected with SARS-CoV-2 diagnosed by RT-PCR on nasopharyngeal swab [Citation47]. Cases were defined as patients needing endotracheal intubation or CPAP/biPAP ventilation. Controls were oligo-asymptomatic subjects not requiring hospitalization.
Ethics approval
The GEN-COVID study was approved by the University Hospital of Siena Ethical Review Board (Protocol n. 16,929, dated 16 March 2020).
LASSO logistic regression
We adopted the Least Absolute Shrinkage and Selection Operator (LASSO) logistic regression model for the classification of severe COVID-19 patients (cases) versus SARS-CoV-2 PCR-positive oligo-asymptomatic subjects (controls), able to enforce both the sparsity and the interpretability of the results. By denoting with
the coefficients of the logistic regression and by lambda (λ) the strength of the regularization, the LASSO regularization [Citation48] term of the loss, has the effect of shrinking the estimated coefficients to 0. In this way, the weights of the logistic regression algorithm can be interpreted as the feature importances of the subset of the most relevant features for the task [Citation49]. The input features are the common bi-allelic polymorphisms from whole-exome sequencing as well as gender, and the age, the latter as a continuous variable normalized between 0 and 1. Common bi-allelic polymorphisms are defined as combinations of two polymorphisms, each with minor allele frequency above 1%, with frequency above 5% in the cohort.
The fundamental hyper-parameter of the logistic regression algorithm is the strength of the LASSO term, which is tuned with a grid search method on the average area under the ROC curve for the 10-fold cross-validation. The regularization hyperparameter varies in the range [10−3, 102] with 50 equally spaced values in the logarithmic scale. The optimal regularization parameter is chosen by selecting the parameter with the highest cross-validation score. During the fitting procedure, the class slight unbalancing is tackled by penalizing the misclassification of the minority class with a multiplicative factor inversely proportional to the class frequencies. The data pre-processing was coded in Python, whereas for the logistic regression model the scikit-learn module with the liblinear coordinate descent optimization algorithm was used. Performances of the model were evaluated using the cross-validation confusion matrix as well as by computing precision, sensitivity, specificity, and the ROC curve.
Cell culture and transfection
HEK-Dual™ Null (NF/IL8) cells (Invivogen, hkd-nullni) cells were cultured in Dulbecco modified Eagle medium (DMEM; Euroclone, ECB7501L) supplemented with 10% fetal bovine serum (FBS; Euroclone, ECS0180L), 2 mM L-glutamine (Carlo Erba, ABP379-100) and 100 units/ml penicillin-streptomycin (Life Technologies, 15,140,148) at 37°C in an atmosphere of 5% CO2:air. Transfections were performed with 1 μg DNA plasmid using lipofectamine LTX (Life Technologies, 15,338,500). The cells were seeded to be 70% to 80% confluent, then DNA was diluted in DMEM with 10 mM HEPES, pH 7.2. Lipofectamine LTX was next added to the complex (5 μl) to allow creation of complexes (30 min at RT). Ultimately, DNA-lipid complexes were added to cells. Bafilomycin A1 was from Santa Cruz Biotechnology (sc-201,550). Human primary fibroblasts were obtained from the Genetic Biobank of Siena (http://biobanknetwork.telethon.it/; cell line number: Rett 2250, 2980/18, 1031/15, Rett 1200). Fibroblasts were cultured in Dulbecco Modified Eagle medium supplemented with 10% FBS, 2% L-glutamine and 1% penicillin-streptomycin, according to standard protocols, and routinely passed 1:2 with trypsin-EDTA (0.05%) solution (Irvine Scientific, 9342).
Immunofluorescence (IF)
Cells were washed with phosphate-buffered saline (PBS; Oxoid, BR0014G), then fixed with 4% paraformaldehyde in PBS for 20 min, washed with PBS and permeabilized with digitonin solution (Life Technologies, BN2006) for 20 min. Then, the cells were washed three times in PBS. Permeabilized cells were incubated with anti-LC3B (MBL, M152–3) and/or anti-LAMP1 (Cell Signaling Technology, 9091) primary antibodies for 1 h, washed three times with PBS, and then incubated with anti-mouse Alexa Fluor 488-conjugated (Life Technologies, A21202) and/or Alexa Fluor 647 (Life Technologies, A21245) secondary antibodies; subsequently cells were washed three times with PBS. Nuclei were stained with a solution of 6 μM of 4ʹ,6-diamidino-2-phenylindole (DAPI; Sigma Aldrich, D9542) in PBS for 10 min. Coverslips were mounted in a fluorescence mounting medium (Dako, S3023). Samples were visualized on a TSC SP5 confocal microscope (Leica Microsystems, 5,100,000,750) installed on an inverted LEICA DMI 6000CS (Leica Microsystems, 10,741,320) microscope and equipped with an oil immersion PlanApo 63 × 1.4 NA objective. Images were acquired using the LAS AF acquisition software (Leica Microsystems, 10,210). Poly(I:C) was from InvivoGen (31,852–29-6).
Dot count and statistical analysis for autophagy
For the LC3B-positive dot counts, we performed intensitometric analysis of fluorescence using the Quantitation Module of Volocity software (PerkinElmer Life Science). LC3B-LAMP1 colocalization rate was measured by the Quantification tool of LAS AF software (Leica Microsystems). Dot counts and colocalization rate were subjected to statistical analysis. Measures were obtained by analyzing at least 400 cells/sample (dot counts) or 250 cells/sample (colocalization rate) from 3 different experiments. Significance (P value) was assessed by Student’s t test, using GraphPad Prism6 software. Asterisks were attributed for the following significance values: P > 0.05 (ns), P < 0.05 (*), P < 0.01 (**), P < 0.001 (***).
Real time qPCR analysis of TNF expression
Total RNA was isolated using the RNAeasy Mini Kit (Qiagen, NC9677589) according to the manufacturer’s instructions. cDNA synthesis was performed using the Maxima First Strand cDNA Synthesis Kit (Life Technologies, EP0751). Neo-synthetized cDNA was used to perform Real Time PCR using the PowerUp Sybr Green (Life Technologies, A25779). The following primers were used: TNF Fw CTATCTGGGAGGGGTCTTCC; TNF Rw GGTTGAGGGTGTCTGAAGGA; HPRT1 Fw GTCTTGCTCGAGATGTGATG and HPRT1 Rw GTAATCCAGCAGGTCAGCAA. Target transcripts were analyzed with the QuantStudio 7 System (Applied Biosystems, CA, USA). The comparative threshold cycle (Ct) method was used for quantification analysis. The Ct values of each gene were normalized to the Ct value of HPRT1. The gene expression levels were evaluated by the fold change using the equation 2-ΔΔCt.
HLA sequencing
HLA-class I and II genes were targeted for DNA sequencing using a biotinylated DNA probe-based capture method [Citation50], with modifications as follows. Genomic DNA (500 ng from each sample) was fragmented enzymatically using the NEBNext Ultra ii FS module (New England Biolabs, E7810S). Individual samples were labeled uniquely using 3 μl of 15 μM custom dual-index adapters (Integrated DNA Technologies, Coralville, IA, USA) and the NEB ligation module. Post ligation cleanup was based on the Kapa Hyper Prep protocol (Kapa Biosystems, Wilmington, MA) and followed by dual size selection. Paired ends of 250 bp each were sequenced using a NovaSeq instrument and SP Reagent Kit (Illumina Inc, San Diego, CA, USA). HLA alleles were determined from the sequence data using the consensus from three algorithms: NGSengine 2.10.0 (GenDX, Utrecht, The Netherlands), HLA Twin (Omixon Biocomputing Ltd. Budapest, Hungary) and HLA*LA [Citation51].
Supplemental Material
Download MS Word (4 MB)Acknowledgments
This study is part of the GEN-COVID Multicenter Study, https://sites.google.com/dbm.unisi.it/gen-covid, the Italian multicenter study aimed at identifying the COVID-19 host genetic bases. Specimens were provided by the COVID-19 Biobank of Siena, which is part of the Genetic Biobank of Siena, member of BBMRI-IT, of the Telethon Network of Genetic Biobanks (project no. GTB18001), of EuroBioBank, and of RD-Connect. We thank the CINECA consortium for providing computational resources and the Network for Italian Genomes http://www.nig.cineca.it for its support. We thank private donors for the support provided to A.R. (Department of Medical Biotechnologies, University of Siena) for the COVID-19 host genetics research project (D.L n.18 of 17 March 2020). PJN was supported by a grant from fastgrants.org and US NIH R56 AI151549. We also thank the COVID-19 Host Genetics Initiative (https://www.covid19hg.org/) and MIUR project “Dipartimenti di Eccellenza 2018-2020” to the Department of Medical Biotechnologies University of Siena, Italy. We thank Dr. Margherita Leonardi for the experimental contribution in the autophagy data analysis. TDJF received the Post-Doctoral fellowship from Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) of Brazil.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Supplementary material
Supplemental data for this article can be accessed here.
Additional information
Funding
References
- Lee IH, Lee JW, Kong SW. A survey of genetic variants in SARS-CoV-2 interacting domains of ACE2, TMPRSS2 and TLR3/7/8 across populations. Infect Genet Evol. 2020;85:104507.
- Mukherjee S, Huda S, Sinha Babu SP. Toll-like receptor polymorphism in host immune response to infectious diseases: a review. Scand J Immunol. 2019 Jul;90(1):e12771.
- Perales-Linares R, Navas-Martin S. Toll-like receptor 3 in viral pathogenesis: friend or foe? Immunology. Immunology. 2013 Oct;140(2):153–167.
- Totura AL, Whitmore A, Agnihothram S, et al. Toll-Like receptor 3 signaling via TRIF contributes to a protective innate immune response to severe acute respiratory syndrome coronavirus infection. mBio 2015 May 26;6(3):e00638–15.
- Matsumoto M, Oshiumi H, Seya T. Antiviral responses induced by the TLR3 pathway. Rev Med Virol. 2011 Mar;21(2):67–77.
- Schulz O, Diebold SS, Chen M, et al. Toll-like receptor 3 promotes cross-priming to virus-infected cells. Nature. 2005 Feb 24;433(7028):887–892.
- Suresh MV, Dolgachev VA, Zhang B, et al. TLR3 absence confers increased survival with improved macrophage activity against pneumonia. JCI Insight. 2019 Dec 5;4(23):e131195.
- Ranjith-Kumar CT, Miller W, Sun J, et al. Effects of single nucleotide polymorphisms on Toll-Like receptor 3 activity and expression in cultured cells. J Biol Chem. 2007 Jun 15;282(24):17696–17705.
- Teimouri H, Maali A. Single-nucleotide polymorphisms in host pattern-recognition receptors show association with antiviral responses against SARS-CoV-2, in-silico trial. JoMMID. 2020;8(2):65–70.
- Dhangadamajhi G, Rout R, Cavazos-Escobar E. Association of TLR3 functional variant (rs3775291) with COVID-19 susceptibility and death: a population-scale study. Hum Cell. 2021 Feb 22;34:1–3.
- Franco LH, Fleuri AKA, Pellison NC, et al. Autophagy downstream of endosomal Toll-Like receptor signaling in macrophages is a key mechanism for resistance to Leishmania major infection. J Biol Chem. 2017 Aug 11;292(32):13087–13096.
- Kirkin V, McEwan DG, Novak I, et al. A role for ubiquitin in selective autophagy. Mol Cell. 2009 May 15;34(3):259–269.
- Delgado MA, Elmaoued RA, Davis AS, et al. Toll-like receptors control autophagy. EMBO J. 2008 Apr 9;27(7):1110–1121.
- Levine B, Mizushima N, Virgin HW. Autophagy in immunity and inflammation. Nature. 2011 Jan 20;469(7330):323–335.
- Prentice E, Jerome WG, Yoshimori T, et al. Coronavirus replication complex formation utilizes components of cellular autophagy. J Biol Chem. 2004 Mar 12;279(11):10136–10141.
- Guo L, Yu H, Gu W, et al. Autophagy negatively regulates transmissible gastroenteritis virus replication. Sci Rep. 2016 Mar 31;6: 23864.
- Carvalho-Schneider C, Laurent E, Lemaignen A, et al. Follow-up of adults with noncritical COVID-19 two months after symptom onset. Clin Microbiol Infect. 2021 Feb;27(2):258–263.
- Hoffmann M, Kleine-Weber H, Schroeder S, et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. 2020Apr16;181(2):271–280. e8.
- Miao Y, Fan L, Li JY. Potential treatments for COVID-19 related cytokine storm - beyond corticosteroids. Front Immunol. 2020 Jun 16;11: 1445.
- Shojaei S, Suresh M, Klionsky DJ, et al. Autophagy and SARS-CoV-2 infection: a possible smart targeting of the autophagy pathway. Virulence. 2020 Dec;11(1):805–810.
- Benvenuto D, Angeletti S, Giovanetti M, et al. Evolutionary analysis of SARS-CoV-2: how mutation of non-structural protein 6 (NSP6) could affect viral autophagy. J Infect. 2020 Jul;81(1):e24–e27.
- Jamwal S, Gautam A, Elsworth J, et al. An updated insight into the molecular pathogenesis, secondary complications and potential therapeutics of COVID-19 pandemic. Life Sci. 2020 Sep 15;257: 118105.
- Torcia MG, Nencioni L, Clemente AM, et al. Sex differences in the response to viral infections: TLR8 and TLR9 ligand stimulation induce higher IL10 production in males. PLoS One. 2012;7(6):e39853.
- Lim HK, Huang SXL, Chen J, et al. Severe influenza pneumonitis in children with inherited TLR3 deficiency. J Exp Med. 2019 Sep 2;216(9):2038–2056.
- Zhang Q, Bastard P, Liu Z, et al. Inborn errors of type I IFN immunity in patients with life-threatening COVID-19. Science. 2020 Oct 23;370(6515):eabd4570.
- Enkhbayar P, Kamiya M, Osaki M, et al. Structural principles of leucine-rich repeat (LRR) proteins. Proteins. 2004 Feb 15;54(3):394–403.
- Rothberg JM, Jacobs JR, Goodman CS, et al. slit: an extracellular protein necessary for development of midline glia and commissural axon pathways contains both EGF and LRR domains. Genes Dev. 1990 Dec;4(12A):2169–2187.
- Kobe B, Kajava AV. The leucine-rich repeat as a protein recognition motif. Curr Opin Struct Biol. 2001 Dec;11(6):725–732.
- Gao T, Zhang SP, Wang JF, et al. TLR3 contributes to persistent autophagy and heart failure in mice after myocardial infarction. J Cell Mol Med. 2018 Jan;22(1):395–408.
- Mehra MR, Ruschitzka F, Patel AN. Retraction-Hydroxychloroquine or chloroquine with or without a macrolide for treatment of COVID-19: a multinational registry analysis. Lancet. 2020 Jun 13;395(10240):1820.
- Mahase E. Covid-19: WHO halts hydroxychloroquine trial to review links with increased mortality risk. BMJ 2020 May 28;369: m2126.
- Ayele Mega T, Feyissa TM, Dessalegn Bosho D, et al. The outcome of hydroxychloroquine in patients treated for COVID-19: systematic review and meta-analysis. Can Respir J. 2020 Oct 13;2020: 4312519.
- Klionsky DJ, Abdel-Aziz AK, Abdelfatah S, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (4th edition). Autophagy 2021 Feb;8:1–382.
- Levy JMM, Towers CG, Thorburn A. Targeting autophagy in cancer. Nat Rev Cancer. 2017 Sep;179:528–542. [ Epub 2017 Jul 28]. PMID: 28751651; PMCID: PMC5975367.
- Michalakis K, Ilias I. SARS-CoV-2 infection and obesity: common inflammatory and metabolic aspects. Diabetes Metab Syndr. 2020 Jul-Aug;14(4):469–471.
- Vomero M, Barbati C, Colasanti T, et al. Autophagy modulation in lymphocytes from COVID-19 patients: new therapeutic target in SARS-COV-2 infection. Front Pharmacol. 2020 Nov 19;11: 569849.
- Jang CH, Choi JH, Byun MS, et al. Chloroquine inhibits production of TNF-alpha, IL-1beta and IL-6 from lipopolysaccharide-stimulated human monocytes/macrophages by different modes. Rheumatology (Oxford). 2006 Jun;45(6):703–710.
- Hayn M, Hirschenberger M, Koepke L, et al. Systematic functional analysis of SARS-CoV-2 proteins uncovers viral innate immune antagonists and remaining vulnerabilities. Cell Rep. 2021 May 18;357:109126. [ Epub 2021 Apr 27]. PMID: 33974846; PMCID: PMC8078906.
- Nahum A, Dadiac H, Batesac A, et al. The biological significance of TLR3 variant, L412F, in conferring susceptibility to cutaneous candidiasis, CMV and autoimmunity. Autoimmun Rev. 2012 Mar;11(5):341–347.
- Aslaksen S, Wolff AB, Vigeland MD, et al. Identification and characterization of rare toll-like receptor 3 variants in patients with autoimmune Addison’s disease. J Transl Autoimmun. 2019 May 28;1:100005.
- Wu DJ, Adamopoulos IE. Autophagy and autoimmunity. Clin Immunol. 2017Mar;176:55–62.
- Caza TN, Talaber G, Perl A. Metabolic regulation of organelle homeostasis in lupus T cells. Clin Immunol. 2012 Sep;144(3):200–213.
- Keller CW, Lünemann JD. Autophagy and autophagy-related proteins in CNS autoimmunity. Front Immunol. 2017 Feb 27;8: 165.
- De Sousa E, Ligeiro D, Lérias JR, et al. Mortality in COVID-19 disease patients: correlating the association of major histocompatibility complex (MHC) with severe acute respiratory syndrome 2 (SARS-CoV-2) variants. Int J Infect Dis. 2020 Sep;98:454–459. Epub 2020 Jul 18.
- Erichsen MM, Løvås K, Skinningsrud B, et al. Clinical, immunological, and genetic features of autoimmune primary adrenal insufficiency: observations from a Norwegian registry. J Clin Endocrinol Metab. 2009 Dec;94(12):4882–4890.
- Smigoc Schweiger D, Mendez A, Kunilo Jamnik S, et al. High-risk genotypes HLA-DR3-DQ2/DR3-DQ2 and DR3-DQ2/DR4-DQ8 in co-occurrence of type 1 diabetes and celiac disease. Autoimmunity. 2016 Jun;49(4):240–247.
- Daga S, Fallerini C, Baldassarri M, et al. Employing a systematic approach to biobanking and analyzing clinical and genetic data for advancing COVID-19 research. Eur J Hum Genet. 2021 Jan;17:1–15.
- Molnar R. Interpretable machine learning. A guide for making black box models explainable. 2020. lulu.com.
- Tibshirani R. Regression shrinkage and selection via the Lasso. Journal of the Royal Statistical Society: Series B (Methodological). 1996;58(1):267–288.
- Norman PJ, Hollenbach JA, Nemat-Gorgani N, et al. Defining KIR and HLA Class I genotypes at highest resolution via high-throughput sequencing. Am J Hum Genet. 2016 Aug 4;99(2):375–391.
- Dilthey AT, Mentzer AJ, Carapito R, et al. HLA*LA-HLA typing from linearly projected graph alignments. Bioinformatics. 2019 Nov 1;35(21):4394–4396.
Appendix
GEN-COVID Multicenter Study (https://sites.google.com/dbm.unisi.it/gen-covid)
Mirella Bruttini1,2,17, Rossella Tita17, Sara Amitrano17, Anna Maria Pinto17, Maria Antonietta Mencarelli17, Caterina Lo Rizzo17, Valentina Perticaroli1,2,17, Massimiliano Fabbiani19, Barbara Rossetti19, Giacomo Zanelli2,19, Elena Bargagli20, Laura Bergantini20, Miriana D’Alessandro20, Paolo Cameli20, David Bennett20, Federico Anedda21, Simona Marcantonio21, Sabino Scolletta21, Federico Franchi21, Maria Antonietta Mazzei22, Susanna Guerrini22, Edoardo Conticini23, Luca Cantarini23, Bruno Frediani23, Danilo Tacconi24, Chiara Spertilli24, Marco Feri25, Alice Donati25, Raffaele Scala26, Luca Guidelli26, Genni Spargi27, Marta Corridi27, Cesira Nencioni28, Leonardo Croci28, Gian Piero Caldarelli29, Maurizio Spagnesi30, Paolo Piacentini30, Maria Bandini30, Elena Desanctis30, Silvia Cappelli30, Anna Canaccini31, Agnese Verzuri31, Valentina Anemoli31, Agostino Ognibene32, Alessandro Pancrazi32, Maria Lorubbio32, Massimo Vaghi33, Antonella D’Arminio Monforte34, Esther Merlini34, Federica Gaia Miraglia34, Raffaele Bruno35,36, Marco Vecchia35, Serena Ludovisi37, Massimo Girardis38, Sophie Venturelli38, Marco Sita38, Andrea Cossarizza39, Andrea Antinori40, Alessandra Vergori40, Arianna Emiliozzi40, Stefano Rusconi41,42, Matteo Siano42, Arianna Gabrieli42, Agostino Riva41,42, Daniela Francisci4344, Elisabetta Schiaroli4344, Francesco Paciosi44, Pier Giorgio Scotton45, Francesca Andretta45, Sandro Panese45, Renzo Scaggiante47, Francesca Gatti48, Saverio Giuseppe Parisi48, Melania degli Antoni49, Isabella Zanella51,41, Matteo Della Monica52, Carmelo Piscopo52, Mario Capasso53,54,55, Roberta Russo53,54, Immacolata Andolfo53,54, Achille Iolascon53,54, Giuseppe Fiorentino55, Massimo Carella56, Marco Castori56, Filippo Aucella57, Pamela Raggi58, Carmen Marciano58, Rita Perna58, Matteo Bassetti59,60, Antonio Di Biagio59,60, Maurizio Sanguinetti61,62, Luca Masucci61,62, Serafina Valente63, Marco Mandalà64, Alessia Giorli64, Lorenzo Salerni64, Patrizia Zucchi65, Pierpaolo Parravicini65, Elisabetta Menatti66 Stefano Baratti45, Tullio Trotta67, Ferdinando Giannattasio67, Gabriella Coiro67, Fabio Lena68, Domenico A. Coviello69, Cristina Mussini70, Giancarlo Bosio71, Enrico Martinelli71, Sandro Mancarella72, Luisa Tavecchia72, Mary Ann Belli72, Lia Crotti73,74,75,76, Gianfranco Parati73,74, Marco Gori77,78, Maurizio Sanarico79, Stefano Ceri80, Pietro Pinoli80, Francesco Raimondi81, Filippo Biscarini82, Alessandra Stella82, Marco Rizzi83, Franco Maggiolo83, Diego Ripamonti83, Claudia Suardi84, Tiziana Bachetti85, Maria Teresa La Rovere86, Simona Sarzi-Braga87, Maurizio Bussotti88, Katia Capitani2,89, Kristina Zguro2, Simona Dei90, Sabrina Ravaglia91, Rosangela Artuso92, Antonio Perrella93, Francesco Bianchi2,93, Giuseppe Merla53,94, Gabriella Maria Squeo94, Mario Tumbarello2,19, Ilaria Rancan2,19, Davide Romani30, Manola Pisani31, Stefano Busani38, Andrea Tommasi43, Francesco Castelli49, Eugenia Quiros-Roldan49, Alessandra Guarnaccia61, Oreste De Vivo63, Gabriella Doddato1,2, Annarita Giliberti1,2, Francesca Ariani1,2,17, Gianluca Lacerenza95, Elena Andreucci92, Giulia Gori92, Angelica Pagliazzi92, Erika Fiorentini92, Paola Bergomi96, Emanuele Catena96, Riccardo Colombo96, Sauro Luchi97, Giovanna Morelli97, Paola Petrocelli97, Sarah Iacopini97, Sara Modica97, Silvia Baroni98, Francesco Vladimiro Segala99, Francesco Menichetti100, Marco Falcone100, Giusy Tiseo100, Chiara Barbieri100, Tommaso Matucci100, Davide Grassi101, Claudio Ferri101, Franco Marinangeli102, Francesco Brancati103, Antonella Vincenti104, Valentina Borgo104, Lombardi Stefania104, Mirco Lenzi104, Massimo Antonio Di Pietro105, Francesca Vichi105, Benedetta Romanin105, Letizia Attala105, Cecilia Costa105, Andrea Gabbuti105, Menè Roberto106, Umberto Zuccon107, Lucia Vietri107, Patrizia Casprini108, Marcello Maffezzoni109 and Marta Colaneri110
19Department of Medical Sciences, Infectious and Tropical Diseases Unit, Azienda Ospedaliera Universitaria Senese, Siena, Italy
20Respiratory Diseases Unit, Department of Medicine, Surgery and Neurosciences, Siena University Hospital, Siena, Italy
21Department of Emergency and Urgency, Medicine, Surgery and Neurosciences, Unit of Intensive Care Medicine, Siena University Hospital, Italy
22Department of Medical, Surgical and Neurosciences and Radiological Sciences, Unit of Diagnostic Imaging, University of Siena
23Rheumatology Unit, Department of Medicine, Surgery and Neurosciences, University of Siena, Policlinico Le Scotte, Italy
24Department of Specialized and Internal Medicine, Infectious Diseases Unit, San Donato Hospital Arezzo, Italy
25Department of Emergency, Anesthesia Unit, San Donato Hospital, Arezzo, Italy
26Department of Specialized and Internal Medicine, Pneumology Unit and UTIP, San Donato Hospital, Arezzo, Italy
27Department of Emergency, Anesthesia Unit, Misericordia Hospital, Grosseto, Italy
28Department of Specialized and Internal Medicine, Infectious Diseases Unit, Misericordia Hospital, Grosseto, Italy
29Clinical Chemical Analysis Laboratory, Misericordia Hospital, Grosseto, Italy
30Department of Preventive Medicine, Azienda USL Toscana Sud Est, Italy
31Territorial Scientific Technician Department, Azienda USL Toscana Sud Est, Italy
32Laboratory Medicine Department, San Donato Hospital, Arezzo, Italy
33Chirurgia Vascolare, Ospedale Maggiore di Crema, Italy
34Department of Health Sciences, Clinic of Infectious Diseases, ASST Santi Paolo e Carlo, University of Milan, Italy
35Division of Infectious Diseases and Immunology, Fondazione IRCCS Policlinico San Matteo, Pavia, Italy
36Department of Internal Medicine and Therapeutics, University of Pavia, Italy
37Fondazione IRCCS Ca’ Granda Ospedale Maggiore Policlinico, Milano, Italy
38Department of Anesthesia and Intensive Care, University of Modena and Reggio Emilia, Modena, Italy
39Department of Medical and Surgical Sciences for Children and Adults, University of Modena and Reggio Emilia, Modena, Italy
40HIV/AIDS Department, National Institute for Infectious Diseases, IRCCS, Lazzaro Spallanzani, Rome, Italy
41III Infectious Diseases Unit, ASST-FBF-Sacco, Milan, Italy
42Department of Biomedical and Clinical Sciences Luigi Sacco, University of Milan, Milan, Italy
43Infectious Diseases Clinic, Department of Medicine , University of Perugia, Santa Maria della Misericordia Hospital, Perugia, Italy
44
45Department of Infectious Diseases, Treviso Hospital, Local Health Unit 2 Marca Trevigiana, Treviso, Italy
46Clinical Infectious Diseases, Mestre Hospital, Venezia, Italy.
47Infectious Diseases Clinic, ULSS1, Belluno, Italy
48Department of Molecular Medicine, University of Padova, Italy
49Department of Infectious and Tropical Diseases, University of Brescia and ASST Spedali Civili Hospital, Brescia, Italy
50Department of Molecular and Translational Medicine, University of Brescia, Italy;
51Clinical Chemistry Laboratory, Cytogenetics and Molecular Genetics Section, Diagnostic Department, ASST Spedali Civili di Brescia, Italy
52Medical Genetics and Laboratory of Medical Genetics Unit, A.O.R.N. “Antonio Cardarelli”, Naples, Italy
53Department of Molecular Medicine and Medical Biotechnology, University of Naples Federico II, Naples, Italy
54CEINGE Biotecnologie Avanzate, Naples, Italy
55Unit of Respiratory Physiopathology, AORN dei Colli, Monaldi Hospital, Naples, Italy
56Division of Medical Genetics, Fondazione IRCCS Casa Sollievo della Sofferenza Hospital, San Giovanni Rotondo, Italy
57Department of Medical Sciences, Fondazione IRCCS Casa Sollievo della Sofferenza Hospital, San Giovanni Rotondo, Italy
58Clinical Trial Office, Fondazione IRCCS Casa Sollievo della Sofferenza Hospital, San Giovanni Rotondo, Italy
59Department of Health Sciences, University of Genova, Genova, Italy
60Infectious Diseases Clinic, Policlinico San Martino Hospital, IRCCS for Cancer Research Genova, Italy
61Microbiology, Fondazione Policlinico Universitario Agostino Gemelli IRCCS, Catholic University of Medicine, Rome, Italy
62Department of Laboratory Sciences and Infectious Diseases, Fondazione Policlinico Universitario A. Gemelli IRCCS, Rome, Italy
63Department of Cardiovascular Diseases, University of Siena, Siena, Italy
64Otolaryngology Unit, University of Siena, Italy
65Department of Internal Medicine, ASST Valtellina e Alto Lario, Sondrio, Italy
66Study Coordinator Oncologia Medica e Ufficio Flussi, Sondrio, Italy
67First Aid Department, Luigi Curto Hospital, Polla, Salerno, Italy
68Local Health Unit-Pharmaceutical Department of Grosseto, Toscana Sud Est Local Health Unit, Grosseto, Italy
69U.O.C. Laboratorio di Genetica Umana, IRCCS Istituto G. Gaslini, Genova, Italy.
70Infectious Diseases Clinics, University of Modena and Reggio Emilia, Modena, Italy.
71Department of Respiratory Diseases, Azienda Ospedaliera di Cremona, Cremona, Italy
72U.O.C. Medicina, ASST Nord Milano, Ospedale Bassini, Cinisello Balsamo (MI), Italy
73Istituto Auxologico Italiano, IRCCS, Department of Cardiovascular, Neural and Metabolic Sciences, San Luca Hospital, Milan, Italy
74Department of Medicine and Surgery, University of Milano-Bicocca, Milan, Italy
75Istituto Auxologico Italiano, IRCCS, Center for Cardiac Arrhythmias of Genetic Origin, Milan, Italy
76Istituto Auxologico Italiano, IRCCS, Laboratory of Cardiovascular Genetics, Milan, Italy
77University of Siena, Diism-sailab, Siena, Italy
78University Cote d’Azur, Inria, CNRS, I3S, Maasai
79Independent Data Scientist, Milan, Italy
80Department of Electronics, Information and Bioengineering (DEIB), Politecnico di Milano, Milano, Italy
81Scuola Normale Superiore, Pisa, Italy
82CNR-Consiglio Nazionale delle Ricerche, Istituto di Biologia e Biotecnologia Agraria (IBBA), Milano, Italy
83Unit of Infectious Diseases, ASST Papa Giovanni XXIII Hospital, Bergamo, Italy
84Fondazione per la ricerca Ospedale di Bergamo, Bergamo, Italy
85Direzione Scientifica, Istituti Clinici Scientifici Maugeri IRCCS, Pavia, Italy
86Istituti Clinici Scientifici Maugeri IRCCS, Department of Cardiology, Institute of Montescano, Pavia, Italy
87Istituti Clinici Scientifici Maugeri, IRCCS, Department of Cardiac Rehabilitation, Institute of Tradate (VA), Italy
88Istituti Clinici Scientifici Maugeri IRCCS, Department of Cardiology, Institute of Milan, Milan, Italy
89IRCCS C. Mondino Foundation,Pavia, Italy
90Core Research Laboratory, ISPRO, Florence, Italy
91Health Management, Azienda USL Toscana Sudest, Tuscany, Italy
92Medical Genetics Unit, Meyer Children’s University Hospital, Florence, Italy
93Department of Medicine, Pneumology Unit, Misericordia Hospital, Grosseto, Italy
94Laboratory of Regulatory and Functional Genomics, Fondazione IRCCS Casa Sollievo della Sofferenza, San Giovanni Rotondo (Foggia), Italy
95Department of Pharmaceutical Medicine, Misericordia Hospital, Grosseto, Italy
96Department of Anesthesia and Intensive Care Unit, ASST Fatebenefratelli Sacco, Luigi Sacco Hospital, Polo Universitario, University of Milan, Milan, Italy
97Infectious Disease Unit, Hospital of Lucca, Lucca, Italy
98Department of Diagnostic and Laboratory Medicine, Unity of Chemistry, Biochemistry and Clinical Molecular Biology, Fondazione Policlinico Universitario A. Gemelli IRCCS, Catholic University of the Sacred Heart, Rome, Italy
99Clinic of Infectious Diseases, Catholic University of the Sacred Heart, Rome, Italy
100Department of Clinical and Experimental Medicine, Infectious Diseases Unit, University of Pisa, Pisa, Italy
101Department of Clinical Medicine, Public Health, Life and Environment Sciences, University of L’Aquila, L’Aquila, Italy
102Anesthesiology and Intensive Care, University of L’Aquila, L’Aquila, Italy
103Medical Genetics Unit, Department of Life, Health and Environmental Sciences, University of L’Aquila, L’Aquila, Italy
104Infectious Disease Unit, Hospital of Massa, Massa Carrara, Italy
105Unit of Infectious Diseases, S.M. Annunziata Hospital, Florence, Italy
106Istituto Auxologico Italiano, IRCCS, Department of Cardiovascular, Neural and Metabolic Sciences, San Luca Hospital; Department of Medicine and Surgery, University of Milano-Bicocca, Milan, Italy
107Respiratory Diseases Unit, “Santa Maria degli Angeli” Hospital, Pordenone, Italy
108Laboratory of Clinical Pathology and Immunoallergy, Florence-Prato, Italy
109University of Pavia, Pavia, Italy
110Division of Infectious Diseases I, Fondazione IRCCS Policlinico San Matteo, Pavia, Italy