
ABSTRACT
Selective degradation of protein aggregates by macroautophagy/autophagy is an essential homeostatic process of safeguarding cells from the effects of proteotoxicity. Among the ubiquitin-like proteins, NEDD8 conjugation to misfolded proteins is prominent in stress-induced protein aggregates, albeit the function of neddylation in autophagy is unclear. Here, we report that polyneddylation functions as a post-translational modification for autophagic degradation of proteotoxic-stress induced protein aggregates. We also show that HYPK functions as an autophagy receptor in the polyneddylation-dependent aggrephagy. The scaffolding function of HYPK is facilitated by its C-terminal ubiquitin-associated domain and N-terminal tyrosine-type LC3-interacting region which bind to NEDD8 and LC3 respectively. Both NEDD8 and HYPK are positive modulators of basal and proteotoxicity-induced autophagy, leading to protection of cells from protein aggregates, such as aggregates of mutant HTT exon 1. Thus, we propose an indispensable and additive role of neddylation and HYPK in clearance of protein aggregates by autophagy, resulting in cytoprotective effect during proteotoxic stress.
Abbreviations: ATG5, autophagy related 5; ATG12, autophagy related 12; ATG14, autophagy related 14; BECN1, beclin 1; CBL, casitas B-lineage lymphoma; CBLB, Cbl proto-oncogene B; GABARAP, GABA type A receptor-associated protein; GABARAPL1, GABA type A receptor associated protein like 1; GABARAPL2, GABA type A receptor associated protein like 2; GFP, green fluorescent protein; HTT, huntingtin; HTT97Q exon 1, huntingtin 97-glutamine exon 1; HUWE1, HECT, UBA and WWE domain containing E3 ubiquitin protein ligase 1; HYPK, huntingtin interacting protein K; IgG, immunoglobulin G; IMR-32, Institute for Medical Research-32; KD, knockdown; Kd, dissociation constant; LAMP1, lysosomal associated membrane protein 1; LIR, LC3 interacting region; MAP1LC3/LC3, microtubule associated protein 1 light chain 3; MAP1LC3A/LC3A, microtubule associated protein 1 light chain 3 alpha; MAP1LC3B/LC3B, microtubule associated protein 1 light chain 3 beta; MARK1, microtubule affinity regulating kinase 1; MARK2, microtubule affinity regulating kinase 2; MARK3, microtubule affinity regulating kinase 3; MARK4, microtubule affinity regulating kinase 4; MCF7, Michigan Cancer Foundation-7; MTOR, mechanistic target of rapamycin kinase; NAE1, NEDD8 activating enzyme E1 subunit 1; NBR1, NBR1 autophagy cargo receptor; NEDD8, NEDD8 ubiquitin like modifier; Ni-NTA, nickel-nitrilotriacetic acid; NUB1, negative regulator of ubiquitin like proteins 1; PIK3C3, phosphatidylinositol 3-kinase catalytic subunit type 3; PolyQ, poly-glutamine; PSMD8, proteasome 26S subunit, non-ATPase 8; RAD23A, RAD23 homolog A, nucleotide excision repair protein; RAD23B, RAD23 homolog B, nucleotide excision repair protein; RFP, red fluorescent protein; RPS27A, ribosomal protein S27a; RSC1A1, regulator of solute carriers 1; SNCA, synuclein alpha; SIK1, salt inducible kinase 1; siRNA, small interfering ribonucleic acid; SOD1, superoxide dismutase 1; SPR, surface plasmon resonance; SQSTM1, sequestosome 1; SUMO1, small ubiquitin like modifier 1; TAX1BP1, Tax1 binding protein 1; TDRD3, tudor domain containing 3; TNRC6C, trinucleotide repeat containing adaptor 6C; TOLLIP, toll interacting protein; TUBA, tubulin alpha; TUBB, tubulin beta class I; UBA, ubiquitin-associated; UBA1, ubiquitin like modifier activating enzyme 1; UBA5, ubiquitin like modifier activating enzyme 5; UBAC1, UBA domain containing 1; UBAC2, UBA domain containing 2; UBAP1, ubiquitin associated protein 1; UBAP2, ubiquitin associated protein 2; UBASH3B, ubiquitin associated and SH3 domain containing B; UBD/FAT10, ubiquitin D; UBE2K, ubiquitin conjugating enzyme E2 K; UBLs, ubiquitin-like proteins; UBL7, ubiquitin like 7; UBQLN1, ubiquilin 1; UBQLN2, ubiquilin 2; UBQLN3, ubiquilin 3; UBQLN4, ubiquilin 4; UBXN1, UBX domain protein 1; ULK1, unc-51 like autophagy activating kinase 1; URM1, ubiquitin related modifier 1; USP5, ubiquitin specific peptidase 5; USP13, ubiquitin specific peptidase 13; VPS13D, vacuolar protein sorting 13 homolog D.
Introduction
Citation95–106Proteostasis is a dynamic process that ensures systemic maintenance of quality and quantity of cellular proteome [Citation1,Citation2]. Stress-induced proteins’ misfolding and aggregation are countered by protein quality control systems [Citation3] which comprise a complex set of mechanisms, starting from guided folding and oligomerization of nascent polypeptides by chaperones [Citation4] to ordered elimination of irreparable and nonfunctional proteins by proteasome and autolysosome machineries [Citation5,Citation6]. While the soluble proteins and smaller protein complexes are cleaved by 26S proteasomal system, the phase-separated insoluble protein aggregates are mainly degraded by autophagosomal/lysosomal pathway [Citation7]. Macroautophagy (hereafter referred as autophagy) is a general mechanism that devours cellular materials, such as damaged organelles, protein aggregates, intracellular pathogens etc., in a membranous vesicle, referred as autophagosome, followed by hydrolytic degradation of autophagosomal contents after fusion of autophagosome with lysosome [Citation8]. Initiation of autophagosome formation from phagophore is critically regulated by several protein complexes (MTOR complex, ULK1 complex, PIK3C3/Vps34 complex, etc.) [Citation9], whereas the maturation stage is more spontaneous with the help of MAP1LC3/LC3, BECN1 and different autophagy-related proteins [Citation10].
Destabilization of cellular proteostasis by intrinsic and extrinsic stresses causes formation of heterogenous protein aggregates [Citation11] which are selectively cleared by autophagy, often the process is termed as aggrephagy [Citation7]. Ubiquitination of proteins is the activation signal for degradation of protein aggregates by canonical autophagy pathway [Citation12]. Covalent linkage of ubiquitin with the target proteins results from sequential activation and functioning of E1, E2 and E3 ligases [Citation13]. Lysine-63 (K63) type polyubiquitin-linked protein aggregates are subjected to autophagic clearance [Citation14]. Although ubiquitination is the major and global modulator of aggrephagy, UBLs are also speculated to have contrasting functions in stress-induced autophagy. While SUMO1 suppresses autophagy [Citation15], it is not known if other UBLs have regulatory functions in autophagy. Particularly, understanding the function of NEDD8, which is a closely related protein to ubiquitin, in proteostasis is not well characterized. Conjugation of NEDD8 to its substrate proteins, mostly the CUL (cullin) proteins [Citation16], is mediated by its own set of ligase proteins [Citation17]. Interestingly, neurodegenerative disease-related proteins (stress granule proteins, inclusion body proteins etc.) are also neddylated [Citation18,Citation19], indicating that neddylation of aggregation-prone proteins functions in clearance of protein aggregates. During proteotoxic stress, neddylation also sequesters the vulnerable nuclear proteins into aggregates [Citation20]. Moreover, thermal and redox stresses increase the cellular level of polyneddylated proteins [Citation21]. While such neddylated proteins undergo NUB1-assisted turnover by proteasomal system [Citation22], the autophagy modulatory functions of NEDD8 remain unclear.
Autophagy receptor proteins are integral components which are required for autophagosomal compartmentalization of protein aggregates [Citation23]. Other than binding to ubiquitin or UBLs, autophagy receptors also recruit LC3, leading to further assembly of autophagosomal machinery to phagophores. SQSTM1/p62 [Citation24], TAX1BP1 [Citation25], TOLLIP [Citation26] etc., are known ubiquitination-dependent autophagy receptors that promote aggrephagy. However, the autophagy receptors of other UBLs are not well defined. In this context, function of a member of intrinsically unstructured chaperone-like proteins (IUPs), named HYPK, in autophagy modulation appears promising. HYPK functions in sequestering and reducing aggregates of different cytosolic and nuclear proteins [Citation27,Citation28]. Previous studies from our group have showed that the balance of structural convolution of HYPK is maintained by complex intra- and inter-molecular interactions of its hydrophobic region, low complexity region and disordered nanostructure [Citation29,Citation30]. Earlier studies found a putative acetyltransferase aiding function of HYPK at ribosome [Citation31]. In Caenorhabditis elegans, F13 G3.10/HYPK forms proteasome blocking complexes [Citation32], with a consequence in aging of the organism [Citation33].
In this article, we report the regulatory functions of UBLs, specifically NEDD8, and HYPK in proteotoxic stress-induced aggrephagy. Assays for functional analysis reveal that polyneddylation serves as a post-translational modification mark for clearance of protein aggregates by autophagy. In this pathway, HYPK functions as an autophagy receptor that scaffolds NEDD8 and LC3 by using its UBA domain and LIR. Both neddylation of proteins and HYPK act as aggrephagy inducers by helping autophagosome biogenesis around protein aggregates, such as those arising from defective ribosomal products of puromycin-treated cells and aggregation-prone HTT exon 1-expressing cells.
Results
siRNA screen for ubiquitin-like proteins that modulate autophagic degradation of proteins during proteotoxic stress
The existing and newly synthesized proteins are vulnerable to proteotoxic stress [Citation34]. Chemical induction of intracellular proteotoxicity by puromycin occurs due to premature translation failure [Citation35], resulting in formation of defective ribosomal products [Citation36] that are primarily cleared from cells by autophagic degradation [Citation37]. While it is known that proteotoxic stress-induced protein aggregates are ubiquitinated prior to their autophagic degradation, the role of other UBLs in this process is less understood. Hence, a screening of UBLs can shed more light on their functions in autophagy. In order to conduct the screen of UBLs that drive the autophagic degradation of proteins, we first determined the conditions of puromycin-induce proteotoxicity that increased autophagy flux without causing cell death. Autophagy flux was monitored by a cell assay that measured the tandem fluorescence of RFP-GFP-LC3B (expressed in a stable cell-line of MCF7) in different conditions. This is a standard autophagy flux assay in which RFP-GFP-LC3B shows green (due to GFP) and red (due to RFP) double fluorescence in autophagic vesicles (AVs) and only red fluorescence in autolysosomes (ALs), indicating spatiotemporal progress of formation and maturation of AVs. Treatment of 15 µg/ml puromycin (this concentration of puromycin was used in all experiments unless otherwise stated) to cells significantly increased the number of GFP+ RFP+ LC3B (AVs) and GFP− RFP+ LC3B (ALs) puncta than untreated cells (). Conversion of native LC3B-I to LC3B-II also increased during puromycin treatment (), indicating high autophagy flux. Thus, we chose puromycin as a reproducible inducer of proteotoxic stress and autophagy in cells.
Figure 1. siRNA screen for autophagy modulatory ubiquitin-like proteins in proteotoxic stress. (A) Stable RFP-GFP-LC3B expressing MCF7 cells were untreated or treated with puromycin (15 μg/ml) for 24 h. Left: confocal fluorescence microscopy images of GFP+ RFP+ LC3B autophagosomes and GFP− RFP+ LC3B autolysosomes. Right: quantitative count (mean ± SD) of GFP+ RFP+ LC3B autophagosomes and GFP− RFP+ LC3B autolysosomes; ~ 100 cells analyzed in each sample (* P < 0.05). (B) Representative image of immunoblot of LC3B in lysate of untreated and puromycin-treated MCF7 cells. Densitometric quantification of LC3B-I and LC3B-II bands relative to TUBA of blots (* P < 0.05). (C and D) Stable RFP-GFP-LC3B expressing MCF7 cells transfected with different ubiquitin-like protein-specific siRNAs and subsequent treatment of cells with puromycin for 36 h. (C) Timeline of the screening experiment and confocal fluorescence microscopy images of GFP+ RFP+ LC3B autophagosomes and GFP− RFP+ LC3B autolysosomes. (D) Mean ± SD number of GFP+ RFP+ LC3B autophagosomes and GFP− RFP+ LC3B autolysosomes; ~ 150 cells analyzed in each experiment (* P < 0.05). (E and F) Representative immunoblot of LC3B from lysate of control-siRNA and UBL-specific siRNA-treated MCF7 cells. Densitometric quantification of LC3B-II bands relative to TUBA of blots (* P < 0.05). (G and H) MCF7 cells transfected with UBL-expressing clones. (G) representative confocal immunofluorescence images of LC3B puncta. (H) mean ± SD number of LC3B puncta/cell in different UBL overexpression condition; ~ 150 cells analyzed in each experiment (* P < 0.05). (I and J) Effect of knockdown of UBLs on expression level of autophagy receptor SQSTM1 and autophagic proteins ATG5, ATG12. (I) representative immunoblots of SQSTM and ATG12–ATG5 in lysate of different UBL knockdown cells. (J) densitometric quantification of SQSTM1 bands relative to TUBA in immunoblots. TUBA is the loading control in immunoblots. Scale bar in confocal microscopy images: 5 μm. All the presented microscopy and immunoblot data are representative of at least three independent experiments.
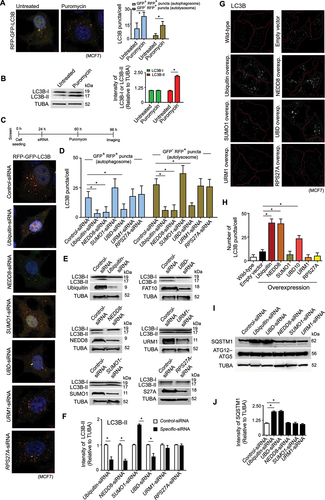
We screened siRNA combinations (6–18 siRNA/gene, 10 nM) against six UBLs, including the ubiquitin-siRNA (positive control), and a negative control-siRNA to understand the potency of the UBLs in modulating autophagy during proteotoxic stress (timeline of screening experiment is in ). Other than ubiquitin-siRNA, the siRNAs targeting NEDD8 and UBD/FAT10 showed repression of autophagy flux. NEDD8-siRNA and UBD-siRNA prevented the formation of GFP+ RFP+ AVs and GFP− RFP+ ALs compared to control-siRNA during proteotoxic stress (, 1D). These two siRNAs also reduced the conversion of LC3B-I to LC3B-II in normal growth condition (, 1 F). Contrary to NEDD8 and UBD/FAT10, downregulation of SUMO1 by siRNA increased formation of AVs, ALs and conversion of LC3B-I to LC3B-II. Thus, NEDD8 and UBD showed autophagy inducing properties like ubiquitin, whereas SUMO1 had opposite effect during proteotoxic stress.
Ubiquitin, NEDD8 and UBD acted as activators of basal autophagy. Higher expression of these three proteins significantly increased autophagy as quantified by the formation of LC3B puncta in cells (, 1H). The effect of NEDD8 in increasing the basal autophagy was similar to ubiquitin but higher than UBD.
Ubiquitin-dependent autophagy involves the receptor function of SQSTM1 [Citation38]. Downregulation of ubiquitin expression caused reduction of autophagy flux and concomitant accumulation of SQSTM1 in cells (, 1 J). While knockdown of UBD showed similar phenotype, NEDD8 downregulation did not show such effect on SQSTM1 levels in cells (, 1 J). Thus, NEDD8 apparently modulated autophagy of proteins in SQSTM1-independent manner, possibly with the involvement of other scaffolding protein(s).
Proteotoxicity-induced NEDD8 granules are cleared by autophagy
The preliminary finding from the knockdown screen that NEDD8 is a positive modulator of autophagy led us to examine the crosstalk between protein neddylation and their autophagic removal during proteotoxic stress. We observed formation of NEDD8 granules in puromycin-treated cells, but not in the untreated cells (). Cell fractionation-derived insoluble cytoplasmic extract contained high molecular weight neddylated protein aggregates (). This observation was in-line with a previous study that also showed similar accumulation of neddylated protein granules during proteotoxic stress [Citation20]. Interestingly, we found that many of the puromycin-induced NEDD8 granules were also positive for LC3B in MCF7 cells (, 2D), as well as in other types (HeLa and IMR-32) of cells (Figure S1A). Chemical inhibition of protein neddylation by MLN4924 (concentration: 1 μM in culture medium of all experiments unless otherwise stated) not only abolished the formation of NEDD8 granules in puromycin-treated cells, but it also depleted the LC3B foci which were otherwise seen at the NEDD8 granules in puromycin-treated cells (). Accordingly, cotreatment of cells with puromycin and MLN4924 decreased the basal level of conversion of LC3B-I to LC3B-II compared to cells that were treated only with puromycin (). These observations indicated that the neddylated protein granules could be undergoing autophagic compartmentalization during proteotoxic stress.
Figure 2. NEDD8 clears proteotoxic stress-induced protein aggregates by autophagy. (A and B) MCF7 cells untreated or treated with puromycin [15 μg/ ml] for 24 h. (A) confocal immunofluorescence microscopy images of NEDD8 granules and mean ± SD quantification of NEDD8 granules; ~ 150 cells analyzed in each sample (* P < 0.05). (B) representative immunoblot of NEDD8 from insoluble and soluble fractions of cell extracts. (C) MCF7 cells were untreated or pretreated with 1 μM MLN4924 for 12 h and then exposed to puromycin [15 μg/ml] for 24 h. Confocal immunofluorescence microscopy images of NEDD8 and LC3B. Quantification of LC3B puncta with mean ± SD in cells; ~ 150 cells analyzed in each sample (* P < 0.05). (D) Representative confocal immunofluorescence microscopy image of colocalization of LC3B with NEDD8 granules in puromycin-treated [15 μg/ml] MCF7 cells. Inset depicts enlarged images of NEDD8 and LC3B positive puncta marked with yellow rectangles. Correlation coefficient of colocalization of NEDD8 granules with LC3B as compared to mCherry in MCF7 cells, ~ 50 cells analyzed in each sample (* P < 0.05). (E) Representative immunoblot of LC3B and NEDD8 for the condition described as in (C), with densitometric quantification of the LC3B-I and LC3B-II bands relative to TUBA of immunoblots (* P < 0.05). (F and G) ATG5, PSMD8 and control knockdown MCF7 cells were treated with puromycin [15 μg/ml], followed by washout of puromycin with normal cell culture medium as presented in timeline of the experiment. (F) confocal immunofluorescence microscopy images of NEDD8 just before and 24 h after puromycin wash. Mean ± SD quantification of NEDD8 granules in different knockdown cells at different timepoints, ~ 150 cells analyzed in each sample (* P < 0.05). Immunoblot of ATG5 and PSMD8 from lysate of control, ATG5 and PSMD8 knockdown cells respectively. (G) Representative immunoblot of NEDD8 from the insoluble fraction of cell lysate of control, ATG5 and PSMD8 knockdown MCF7 cells. (H) Representative immunoblot of autophagy marker proteins from lysate of control and NEDD8 overexpressing MCF7 cells. Densitometric quantification of bands of autophagy marker proteins relative to TUBA of blots (* P < 0.05). (I) Representative immunoblots of phosphoserine 15-BECN1 [p-S15-BECN1] and BECN1 in a time-chase experiment from lysate of control and NEDD8 knockdown MCF7 cells. Densitometric quantification of p-S15-BECN1 relative to BECN1 of the immunoblots (* P < 0.05). TUBA is the loading control in immunoblots. Scale bars in confocal microscopy images: 5 μm, and 1 μm for enlarged images. All the presented microscopy and immunoblot data are representative of at least three independent experiments.
![Figure 2. NEDD8 clears proteotoxic stress-induced protein aggregates by autophagy. (A and B) MCF7 cells untreated or treated with puromycin [15 μg/ ml] for 24 h. (A) confocal immunofluorescence microscopy images of NEDD8 granules and mean ± SD quantification of NEDD8 granules; ~ 150 cells analyzed in each sample (* P < 0.05). (B) representative immunoblot of NEDD8 from insoluble and soluble fractions of cell extracts. (C) MCF7 cells were untreated or pretreated with 1 μM MLN4924 for 12 h and then exposed to puromycin [15 μg/ml] for 24 h. Confocal immunofluorescence microscopy images of NEDD8 and LC3B. Quantification of LC3B puncta with mean ± SD in cells; ~ 150 cells analyzed in each sample (* P < 0.05). (D) Representative confocal immunofluorescence microscopy image of colocalization of LC3B with NEDD8 granules in puromycin-treated [15 μg/ml] MCF7 cells. Inset depicts enlarged images of NEDD8 and LC3B positive puncta marked with yellow rectangles. Correlation coefficient of colocalization of NEDD8 granules with LC3B as compared to mCherry in MCF7 cells, ~ 50 cells analyzed in each sample (* P < 0.05). (E) Representative immunoblot of LC3B and NEDD8 for the condition described as in (C), with densitometric quantification of the LC3B-I and LC3B-II bands relative to TUBA of immunoblots (* P < 0.05). (F and G) ATG5, PSMD8 and control knockdown MCF7 cells were treated with puromycin [15 μg/ml], followed by washout of puromycin with normal cell culture medium as presented in timeline of the experiment. (F) confocal immunofluorescence microscopy images of NEDD8 just before and 24 h after puromycin wash. Mean ± SD quantification of NEDD8 granules in different knockdown cells at different timepoints, ~ 150 cells analyzed in each sample (* P < 0.05). Immunoblot of ATG5 and PSMD8 from lysate of control, ATG5 and PSMD8 knockdown cells respectively. (G) Representative immunoblot of NEDD8 from the insoluble fraction of cell lysate of control, ATG5 and PSMD8 knockdown MCF7 cells. (H) Representative immunoblot of autophagy marker proteins from lysate of control and NEDD8 overexpressing MCF7 cells. Densitometric quantification of bands of autophagy marker proteins relative to TUBA of blots (* P < 0.05). (I) Representative immunoblots of phosphoserine 15-BECN1 [p-S15-BECN1] and BECN1 in a time-chase experiment from lysate of control and NEDD8 knockdown MCF7 cells. Densitometric quantification of p-S15-BECN1 relative to BECN1 of the immunoblots (* P < 0.05). TUBA is the loading control in immunoblots. Scale bars in confocal microscopy images: 5 μm, and 1 μm for enlarged images. All the presented microscopy and immunoblot data are representative of at least three independent experiments.](/cms/asset/a6210b97-3070-49ea-9e0c-57cfd2610652/kaup_a_1997053_f0002_oc.jpg)
Next, we examined if the neddylated granules were cleared through autophagy by knocking down one or more proteins of core autophagy machinery. Although puromycin initiated the accumulation of NEDD8 granules, they were effectively cleared in control knockdown cells after washout of puromycin (, 2G). On the contrary, a significant number of NEDD8 granules persisted in the ATG5 knockdown cells after puromycin washout (, 2 G). Blocking the 26S proteasomal pathway by knockdown of an essential proteasomal protein, PSMD8, also showed accumulation of NEDD8 granules, even though almost five times lesser than the effect of ATG5 knockdown (, 2G). Taken together, these data suggest that proteotoxic stress causes formation of neddylated protein granules that are cleared by autophagy pathway.
To confirm the role of NEDD8 in autophagy, we further studied the expression of autophagy markers during differential expression of NEDD8. Higher expression of NEDD8 in MCF7 cells caused increased lipidation of LC3B-I to LC3B-II, signifying the activation of autophagy pathway (). In addition to MCF7 cells, knock down of NEDD8 in HeLa and IMR-32 cells caused repression of autophagy which was represented by reduced lipidation of LC3B (Figure S1B) and less number of LC3B-positive puncta (Figure S1C) compared to control cells.
Phosphorylation of serine 15 of BECN1 by ULK 1 induces activation of ATG14-bound PIK3C3/VPS34 in nutrient-deficient conditions [Citation39]. Because this is an accelerating event in autophagy, reduction of phosphoserine-15-BECN1 (p-S15-BECN1) would implicate inhibition of autophagy. Similar to the conditions of nutrient stress, we found that proteotoxic stress by puromycin also increases the cellular level of p-S15-BECN1, confirming that proteotoxic stress stimulates autophagy initiation and flux. However, knockdown of NEDD8 temporally reduced the cellular level of p-S15-BECN1 (). Such result again provides the evidence that NEDD8 augments a noncanonical autophagy pathway to degrade proteins.
Polyneddylated aggregation-prone HTT exon 1 is degraded by autophagy
Having found that proteotoxicity-induced NEDD8 granules are cleared by autophagy, we were interested to find if polyneddylated proteins are in general subjected to autophagic degradation. HTT is one of the few number of substrate proteins that undergo polyneddylation [Citation40]. Consistent with the previous studies, we observed that the polyglutamine-expanded mutant HTT exon 1 (HTT97Q exon 1) was polyneddylated (). Denaturing (with 1% SDS) immunoprecipitation of NEDD8 (to purify only neddylated proteins and prevent purification of proteins that are noncovalently associated with NEDD8) followed by immunoblotting against HTT97Q exon 1 showed the high molecular weight neddylated HTT97Q exon 1 (). Neddylation of HTT97Q exon 1 aggregates in IMR-32 and SH-SY5Y (human neuroblastoma) cells was also evident from the observation of high NEDD8 colocalization with HTT97Q exon 1 aggregates (). Since a nonspecific protein (blue fluorescent protein, BFP) did not colocalize with HTT97Q exon 1 (), it was apparent that neddylation of HTT97Q exon 1 was specific, and not just trapping of NEDD8 with the sticky HTT exon 1 aggregates.
Figure 3. Neddylation facilitates aggrephagy of mutant HTT exon 1 aggregates. (A) Representative immunoblots of HTT97Q exon 1 [polyQ] and NEDD8 from denaturing immunoprecipitation of neddylated proteins of extract of stable HTT97Q exon 1 expressing IMR-32 cells transfected with control or NEDD8 siRNAs. (B) Confocal immunofluorescence images of colocalization of NEDD8 with HTT97Q exon 1-GFP in – IMR-32 cells stably expressing HTT97Q exon 1-GFP, – in SH-SY5Y cells transiently transfected with HTT97Q exon 1-GFP. Quantification of colocalization coefficient of HTT97Q exon 1-GFP with NEDD8 compared to BFP, ~ 200 cells analyzed in each sample (* P < 0.05). (C) Representative immunoblot of HTT97Q exon 1 and NEDD8 from lysate of HTT97Q exon 1-expressing IMR-32 cells transfected with control or NEDD8-siRNAs. Densitometric quantification of HTT97Q exon 1 bands relative to TUBA of blots (* P < 0.05). (D and E) Control or NEDD8-siRNA was transfected to stable HTT97Q exon 1-GFP expressing IMR-32 cells. NEDD8 knockdown cells were exposed to 5 μM MG132 or 1 μM bafilomycin A1 for 24 h. (D) confocal fluorescence microscopy images of HTT97Q exon 1-GFP. Immunoblot of NEDD8 from lysate of control and NEDD8 knockdown cells. (E) quantification (mean ± SD) of HTT97Q exon 1-GFP aggregates, ~ 200 cells analyzed in each sample (* P < 0.05). (F) Representative immunoblot of HTT97Q exon 1 and NEDD8 from experiments following the same procedure as in (D), except that the IMR-32 cells stably expressed HTT97Q exon 1. Densitometric quantification of HTT97Q exon 1 bands relative to TUBA of blots (* P < 0.05). (G) Control or ATG5-siRNA was transfected to HTT97Q exon 1 and NEDD8 overexpressing IMR-32 cells in presence of 5 μM MG132. Representative immunoblots of HTT97Q exon 1 and NEDD8. Densitometric quantification of HTT97Q exon 1 bands relative to TUBA of blots (* P < 0.05). TUBA is the loading control in immunoblots. Scale bars in confocal microscopy images: 5 μm. All the presented microscopy and immunoblot data are representative of at least three independent experiments.
![Figure 3. Neddylation facilitates aggrephagy of mutant HTT exon 1 aggregates. (A) Representative immunoblots of HTT97Q exon 1 [polyQ] and NEDD8 from denaturing immunoprecipitation of neddylated proteins of extract of stable HTT97Q exon 1 expressing IMR-32 cells transfected with control or NEDD8 siRNAs. (B) Confocal immunofluorescence images of colocalization of NEDD8 with HTT97Q exon 1-GFP in – IMR-32 cells stably expressing HTT97Q exon 1-GFP, – in SH-SY5Y cells transiently transfected with HTT97Q exon 1-GFP. Quantification of colocalization coefficient of HTT97Q exon 1-GFP with NEDD8 compared to BFP, ~ 200 cells analyzed in each sample (* P < 0.05). (C) Representative immunoblot of HTT97Q exon 1 and NEDD8 from lysate of HTT97Q exon 1-expressing IMR-32 cells transfected with control or NEDD8-siRNAs. Densitometric quantification of HTT97Q exon 1 bands relative to TUBA of blots (* P < 0.05). (D and E) Control or NEDD8-siRNA was transfected to stable HTT97Q exon 1-GFP expressing IMR-32 cells. NEDD8 knockdown cells were exposed to 5 μM MG132 or 1 μM bafilomycin A1 for 24 h. (D) confocal fluorescence microscopy images of HTT97Q exon 1-GFP. Immunoblot of NEDD8 from lysate of control and NEDD8 knockdown cells. (E) quantification (mean ± SD) of HTT97Q exon 1-GFP aggregates, ~ 200 cells analyzed in each sample (* P < 0.05). (F) Representative immunoblot of HTT97Q exon 1 and NEDD8 from experiments following the same procedure as in (D), except that the IMR-32 cells stably expressed HTT97Q exon 1. Densitometric quantification of HTT97Q exon 1 bands relative to TUBA of blots (* P < 0.05). (G) Control or ATG5-siRNA was transfected to HTT97Q exon 1 and NEDD8 overexpressing IMR-32 cells in presence of 5 μM MG132. Representative immunoblots of HTT97Q exon 1 and NEDD8. Densitometric quantification of HTT97Q exon 1 bands relative to TUBA of blots (* P < 0.05). TUBA is the loading control in immunoblots. Scale bars in confocal microscopy images: 5 μm. All the presented microscopy and immunoblot data are representative of at least three independent experiments.](/cms/asset/23b11d6f-bad3-48c9-bfb7-12927ccbef03/kaup_a_1997053_f0003_oc.jpg)
In theory, HTT exon 1 could be subjected to three different types of neddylation – mononeddylation, polyneddylation and multi-mononeddylation. To understand the neddylation pattern of HTT97Q exon 1, we generated a NEDD8 mutant construct in which all the lysine residues of NEDD8 were mutated to arginine (NEDD8-allR) (Figure S2A). This NEDD8-allR mutant cannot covalently attach to another NEDD8 or NEDD8-allR molecule through lysine-linkage, thereby preventing the formation of polyneddylated chains of NEDD8-allR. However, conjugation of NEDD8-allR with proteins can form mononeddylated and multi-mononeddylated substrates. Coexpression of HTT97Q exon 1 with NEDD8 or NEDD8-allR showed that high molecular weight conjugated complexes of HTT97Q exon 1 formed only in presence of NEDD8 (Figure S2B). NEDD8-allR conjugation resulted in mononeddylated HTT97Q exon 1, but not the polyneddylated or multi-mononeddylated HTT97Q exon 1.
During higher expression of NEDD8, thermal stress and proteasomal inhibition, polyubiquitin chains also incorporate NEDD8 (i.e., atypical neddylation forming ubiquitin-NEDD8 hybrid chain) [Citation41]. This phenomenon does not depend upon the NAE1. Instead, NEDD8 is activated by the UBA1 [Citation42]. Unlike thermal stress and proteasome inhibition, we did not observe atypical neddylation of proteins during puromycin-induced proteotoxicity (Figure S3A). NEDD8 immunoprecipitation in denaturing condition from lysate of untreated and puromycin-treated MCF7 cells did not show presence of ubiquitin with NEDD8. Absence of ubiquitin in the NEDD8 affinity-isolation fraction showed that the poly-NEDD8 chains purely contained NEDD8, and they were not hybrid NEDD8-ubiquitin conjugates. We also checked the colocalization of NEDD8 with ubiquitin in untreated and puromycin-treated cells. Ubiquitin did not colocalize with NEDD8 granules in either of the cells (Figure S3B). In addition to this experiment, we analyzed the homogeneity of polyneddylation of HTT97Q exon 1 to clarify if HTT97Q exon 1 was simultaneously modified by NEDD8 and ubiquitin. HTT97Q exon 1 and ubiquitin were immunoblotted from the NEDD8 immunoprecipitated samples. Denaturing immunoprecipitation of NEDD8 from the insoluble fractions of lysate of normal and NEDD8 overexpressing cells showed that HTT97Q exon 1 was polyneddylated, whereas the poly-NEDD8 chains linked to HTT97Q exon 1 lacked any ubiquitin (Figure S4A). This signified that the HTT97Q exon 1-attached poly-NEDD8 chains were comprised of only NEDD8 molecules without any contamination of ubiquitin. A confirmatory test to characterize the HTT97Q exon 1-attached neddylation chains was also done by using NAE1 and UBA1 inhibitors. Treatment of the HTT97Q exon 1 expressing IMR-32 cells with NAE1 inhibitor, MLN4924, decreased polyneddylation of the protein (Figure S4B). Application of UBA1 inhibitor, MLN7243 (concentration: 1 μM in culture medium), to the cells had no effect on the polyneddylation of HTT exon 1 (Figure S4B). Cumulatively, these observations corroborated with the fact that puromycin-induced proteotoxicity results in homogeneous polyneddylation of proteins.
To understand the effect of neddylation on mutant HTT exon 1 degradation by autophagy, we measured the cellular level of HTT97Q exon 1 in varying expression conditions of NEDD8. Knockdown of NEDD8 effectively decreased the degradation of HTT97Q exon 1 compared to control cells (). Number of HTT97Q exon 1 aggregates also increased in the NEDD8-KD cells (, 3E). Exposure of HTT97Q exon 1 expressing cells (in NEDD8-KD background) with proteasomal inhibitor (MG132; concentration: 5 μM in culture medium of all experiments unless otherwise stated) or autophagy inhibitor (bafilomycin A1; concentration: 1 μM in culture medium of all experiments unless otherwise stated) showed further increase of total HTT97Q exon 1 protein and its aggregates (). In this respect, blocking the autophagy pathway had a more pronounced effect than blocking the proteasomal pathway. Previous reports found that neddylated HTT could be cleared through proteasomal pathway by the activity of NUB1 protein [Citation40], possibly aided by the activity of VCP/p97-UFD1-NPLOC4/NPL4 complex [Citation43]. However, given the condition that polyubiquitinated proteins, such as polyubiquitinated HTT, could be redundantly degraded by both proteasomal and autophagosomal pathways [Citation44], we tested if polyneddylated HTT97Q exon 1 was also subjected to autophagosomal degradation. While higher expression of NEDD8 enhanced the clearance of HTT97Q exon 1 from cells, downregulation of ATG5 prevented such NEDD8-facilitated clearance of HTT97Q exon 1 (). In this experiment, MG132 treatment to cells ensured that neddylation-dependent degradation of HTT97Q exon 1 was not occurring through proteasomal pathway. Since NEDD8 could assist the degradation of HTT97Q exon 1 in presence of MG132, it was reasonable to understand that NEDD8 had positive functions in the autophagic degradation of mutant HTT exon 1.
Overall, these observations showed that NEDD8 is an autophagy modulator protein, and polyneddylation serves as a signal for autophagic degradation of protein aggregates.
HYPK is the receptor in neddylation-dependent autophagy
Canonical degradation of polyubiquitinated proteins by autophagy requires the scaffolding function of SQSTM1 protein. SQSTM1 has UBA domain and LIR which are used by the protein to interact with ubiquitin and LC3 respectively. We expected that a protein with similar scaffolding function would be required for the autophagy of polyneddylated proteins. The finding that cellular level of SQSTM1 was unchanged during inhibition of neddylation-dependent autophagy (described in the first section of results; , 1J), led us to turn our attention to identify the scaffolding protein which is involved in simultaneous interactions with NEDD8 and LC3.
(I) HYPK interacts with NEDD8 by using its C-terminal UBA domain
Because UBA domains recognize ubiquitin and UBLs [Citation45], we tested a knockdown screen to understand the effects of thirty UBA domain-containing proteins on accumulation of cellular NEDD8 granules. Downregulation of five UBA domain-containing proteins significantly increased the number of polyneddylated substrate granules in cells (, 4B); with HYPK knockdown showing highest accumulation of neddylated granules (). To get further idea of an ideal binding of NEDD8 with the UBA domains, we conducted computational docking/binding studies of NEDD8 with different UBA domains of selected proteins. A higher free energy of assembly dissociation [ΔG(diss)] would indicate stronger binding of NEDD8 to the respective UBA domain. The binding affinity of one of the UBA domains of NUB1 for NEDD8 was highest, followed by the binding affinity of HYPK-UBA for NEDD8 (), implying that UBA domains of NUB1 and HYPK form thermodynamically stable complexes with NEDD8.
Figure 4. HYPK binds to NEDD8 by using its UBA domain. (A and B) MCF7 cells were transfected with control-siRNA or siRNAs against different UBA domain containing proteins. (A) number (mean ± SD) of NEDD8 granules in cells, ~ 300 cells analyzed in each sample (* P < 0.05). B: Representative confocal immunofluorescence microscopy images of NEDD8. (C) In silico molecular docking of NEDD8 was done on UBA domains of selected human proteins. Free energy of dissociation of assemblies/complexes [ΔG(diss)] was computed in PISA webserver. (D) Representative immunoblots in non-denaturing conditions of – Left: NEDD8, HYPK and FLAG-tagged HYPK-UBA from immunoprecipitated NEDD8 of cell lysate of MCF7 cells [FLAG-tagged HYPK-UBA was ectopically expressed]; Right: HYPK and NEDD8 from immunoprecipitated HYPK of MCF7 cell lysate. (E) Confocal immunofluorescence microscopy images of colocalization of NEDD8, HYPK and FLAG-tagged HYPK-UBA in MCF7 cells. (F) Representation of HYPK, its UBA domain and different mutants of HYPK-UBA domain that were used in the study. HYPK-UBA domain contained the C-terminal 45 amino acid region; the D94 and E101 residues were mutated to alanine in HYPK-UBA D94A, E101A construct; the L113 and G118 residues were deleted in HYPK-UBA [ΔL113, ΔG118] construct. (G) Quantitative binding responses and affinities of interactions between recombinant HYPK, HYPK-UBA, HYPK-UBA D94A, E101A, HYPK-UBA [ΔL113, ΔG118] and NEDD8 as probed by surface plasmon resonance assays. Dissociation constants [Kd values] are given for the interactions. (H) Structural basis and thermodynamic parameters of interactions between NEDD8 and HYPK-UBA in the HYPK-NEDD8 complex. Left: predicted structure of the complex of HYPK and NEDD8. Right: the conserved residues of HYPK-UBA and their cognate interacting residues in NEDD8 are shown with stick representation. Hydrogen bonds are highlighted with black dotted lines. TUBA is the loading control in immunoblots. Scale bars in confocal microscopy images: 5 μm. All the presented microscopy and immunoblot data are representative of at least three independent experiments.
![Figure 4. HYPK binds to NEDD8 by using its UBA domain. (A and B) MCF7 cells were transfected with control-siRNA or siRNAs against different UBA domain containing proteins. (A) number (mean ± SD) of NEDD8 granules in cells, ~ 300 cells analyzed in each sample (* P < 0.05). B: Representative confocal immunofluorescence microscopy images of NEDD8. (C) In silico molecular docking of NEDD8 was done on UBA domains of selected human proteins. Free energy of dissociation of assemblies/complexes [ΔG(diss)] was computed in PISA webserver. (D) Representative immunoblots in non-denaturing conditions of – Left: NEDD8, HYPK and FLAG-tagged HYPK-UBA from immunoprecipitated NEDD8 of cell lysate of MCF7 cells [FLAG-tagged HYPK-UBA was ectopically expressed]; Right: HYPK and NEDD8 from immunoprecipitated HYPK of MCF7 cell lysate. (E) Confocal immunofluorescence microscopy images of colocalization of NEDD8, HYPK and FLAG-tagged HYPK-UBA in MCF7 cells. (F) Representation of HYPK, its UBA domain and different mutants of HYPK-UBA domain that were used in the study. HYPK-UBA domain contained the C-terminal 45 amino acid region; the D94 and E101 residues were mutated to alanine in HYPK-UBA D94A, E101A construct; the L113 and G118 residues were deleted in HYPK-UBA [ΔL113, ΔG118] construct. (G) Quantitative binding responses and affinities of interactions between recombinant HYPK, HYPK-UBA, HYPK-UBA D94A, E101A, HYPK-UBA [ΔL113, ΔG118] and NEDD8 as probed by surface plasmon resonance assays. Dissociation constants [Kd values] are given for the interactions. (H) Structural basis and thermodynamic parameters of interactions between NEDD8 and HYPK-UBA in the HYPK-NEDD8 complex. Left: predicted structure of the complex of HYPK and NEDD8. Right: the conserved residues of HYPK-UBA and their cognate interacting residues in NEDD8 are shown with stick representation. Hydrogen bonds are highlighted with black dotted lines. TUBA is the loading control in immunoblots. Scale bars in confocal microscopy images: 5 μm. All the presented microscopy and immunoblot data are representative of at least three independent experiments.](/cms/asset/c6b2d2f0-b05e-48b2-9570-dc83afcb84a1/kaup_a_1997053_f0004_oc.jpg)
Functional binding of the NUB1-UBA to NEDD8 [Citation46] is known to be involved in NUB1-facilitated degradation of neddylated proteins through proteasomal pathway. On the other hand, aggregate-sequestering function of HYPK is linked to the degradation of the aggregation-prone proteins in an unknown pathway [Citation27]. Thus, it was befitting to understand if HYPK delivers the protein aggregates to autophagic degradation system.
The UBA domain is in the C-terminal region of HYPK [Citation30]. This region is conserved in the HYPK proteins of different organisms (Figure S5A). A multiple sequence alignment-based phylogenetic clustering of similar UBA domains of human proteins led to the observation that the sequence of the second UBA domain (UBA2) of NUB1 protein is closest to the sequence of HYPK-UBA domain (Figure S5B). Thus, we tested if HYPK is a NEDD8 interacting protein.
HYPK displayed strong binding to NEDD8 by using its UBA domain in vitro conditions. Non-denaturing immunoprecipitation of NEDD8 from MCF7 cell lysate, followed by immunoblotting showed that both HYPK and HYPK-UBA could be pulled-down with NEDD8 (). A reciprocal immunoprecipitation of HYPK, followed by immunoblotting for NEDD8 and HYPK was also done to test which forms of NEDD8 bind to HYPK. The immunoblot profile showed interaction of HYPK with monomeric NEDD8 and polyneddylated chains (). We also observed that HYPK and HYPK-UBA signals disappeared in the blots of denaturing immunoprecipitation of NEDD8 (Figure S5C), indicating that HYPK and HYPK-UBA were not themselves neddylated, but they were noncovalently bound to NEDD8. HYPK and HYPK-UBA were also observed to show high spatial colocalization with intracellular NEDD8 of MCF7 and IMR-32 cells (, Figure S6A).
We identified a set of critical residues [aspartate-94 (D94), glutamate-101 (E101), leucine-113 (L113) and glycine-118 (G118); residues were numbered according to their positions in human HYPK isoform-1; NCBI accession number: NP_057484.3] that were required for strong and efficient binding of HYPK to NEDD8. These residues are typically conserved in the HYPK proteins of different organisms (Figure S5A). In order to understand the function of these amino acids in HYPK binding to NEDD8, we generated different mutants of HYPK-UBA in which the conserved residues were either mutated or deleted (). In the HYPK-UBAD94A, E101A construct, the D94 and E101 residues were mutated to alanine, whereas L113 and G118 residues were deleted in HYPK-UBA [ΔL113, ΔG118]. In the protein-protein interaction assays by SPR, both mutants displayed lower but not complete loss of binding affinity for NEDD8 compared to the affinities of wild-type HYPK and HYPK-UBA for NEDD8 (). It implied that the four conserved residues of HYPK-UBA were necessary, but not sufficient, for HYPK-UBA interaction with NEDD8. Although the conserved amino acids in HYPK-UBA were critical for HYPK binding to NEDD8, the neighboring residues possibly provided additional support to the interaction. We also noted that the full-length HYPK protein showed lower binding affinity for NEDD8 than the binding affinity of HYPK-UBA for NEDD8. The reduced binding affinity of HYPK for NEDD8 could be due to the intramolecular interactions between the disorder nanostructure and low complexity region (LCR) of HYPK, as shown in one of our previous studies [Citation29]. Such an intramolecular interaction could possibly interfere in the optimal binding of HYPK-UBA to NEDD8.
In the available crystal structure of HYPK [Citation47], E101 and G118 are surface residues that are exposed to external environment, allowing these two residues to directly interact with the proximal residues of NEDD8 in the model HYPK-NEDD8 complex (). On the other hand, D94 and L113 residues are located at the interface of two helices of the globular core of HYPK-UBA domain (). Thus, these residues are more likely to be involved in stabilization of the HYPK-UBA domain, rather than making direct interactions with NEDD8.
(II) HYPK is an LC3 interacting protein
To understand if HYPK is involved in facilitating the autophagic degradation of neddylated proteins, we investigated the potential interaction of HYPK with LC3. HYPK lacks any conventional LIR which could be represented by [W/Y/F]xx[L/V/I] sequence. However, an unbiased analysis of HYPK sequence predicted a putative atypical LIR sequence represented by Y49AEE52 (). Such an atypical LIR with an aromatic amino acid at the first position and an acidic amino acid (instead of a hydrophobic amino acid) at the fourth position is previously reported in the UBA5 protein (in the UBA5, the LIR is WGIE) [Citation48]. The tyrosine-49 (Y49) is also conserved in the HYPK protein of different organisms ().
Figure 5. HYPK is a LC3 binding protein. (A) Multiple sequence alignment of the putative LC3 interacting region sequences of HYPK of different organisms. (B) Representative immunoblots of HYPK and LC3B from the non-denaturing immunocomplexes of endogenous LC3B of MCF7 cell lysate. (C) Top: schematic representation of cloning of LC3A, LC3B, GABARAP, GABARAPL1, GABARAPL2 and HYPK in pETDuet-1 vector. Bottom: protein-protein interaction analysis by Ni-NTA binding of recombinant 6xhistidin-tagged LC3A, LC3B, GABARAP, GABARAPL1, GABARAPL2 and untagged HYPK coexpressed in BL21DE3 strain of Escherichia coli from pETDuet-1 vector. Immunoblot of HYPK and coomassie blue staining of interacting pairs; empty vector was used as negative control. (D) Representation of HYPK and its mutants used in the study. HYPK-UBA was the C-terminal 45 residue region; HYPK-N84 was the N-terminal 84 amino acid region; the 49th, 51st and 52nd residues of HYPK were mutated to alanine in HYPK Y49AEE52> A49AAA52. (E) Quantitative analysis of binding interaction and affinities between recombinant HYPK, HYPK-UBA, HYPK-N84, HYPK Y49AEE52> A49AAA52 and LC3B as determined by surface plasmon resonance assays. Dissociation constants [Kd values] of bindings are given. (F) MCF7 cells were transfected with HYPK-UBA, HYPK-N84, HYPK Y49AEE52> A49AAA52 [all with N-terminal FLAG tag]. Confocal immunofluorescence microscopy images of HYPK, HYPK-UBA, HYPK-N84 and HYPK Y49AEE52> A49AAA52 and LC3B. Quantitative estimation of colocalization coefficients of proteins; ~ 200 cells analyzed in each sample. TUBA is the loading control in immunoblots. Scale bars in confocal microscopy images represent 5 μm. All the presented microscopy and immunoblot data are representative of at least three independent experiments.
![Figure 5. HYPK is a LC3 binding protein. (A) Multiple sequence alignment of the putative LC3 interacting region sequences of HYPK of different organisms. (B) Representative immunoblots of HYPK and LC3B from the non-denaturing immunocomplexes of endogenous LC3B of MCF7 cell lysate. (C) Top: schematic representation of cloning of LC3A, LC3B, GABARAP, GABARAPL1, GABARAPL2 and HYPK in pETDuet-1 vector. Bottom: protein-protein interaction analysis by Ni-NTA binding of recombinant 6xhistidin-tagged LC3A, LC3B, GABARAP, GABARAPL1, GABARAPL2 and untagged HYPK coexpressed in BL21DE3 strain of Escherichia coli from pETDuet-1 vector. Immunoblot of HYPK and coomassie blue staining of interacting pairs; empty vector was used as negative control. (D) Representation of HYPK and its mutants used in the study. HYPK-UBA was the C-terminal 45 residue region; HYPK-N84 was the N-terminal 84 amino acid region; the 49th, 51st and 52nd residues of HYPK were mutated to alanine in HYPK Y49AEE52> A49AAA52. (E) Quantitative analysis of binding interaction and affinities between recombinant HYPK, HYPK-UBA, HYPK-N84, HYPK Y49AEE52> A49AAA52 and LC3B as determined by surface plasmon resonance assays. Dissociation constants [Kd values] of bindings are given. (F) MCF7 cells were transfected with HYPK-UBA, HYPK-N84, HYPK Y49AEE52> A49AAA52 [all with N-terminal FLAG tag]. Confocal immunofluorescence microscopy images of HYPK, HYPK-UBA, HYPK-N84 and HYPK Y49AEE52> A49AAA52 and LC3B. Quantitative estimation of colocalization coefficients of proteins; ~ 200 cells analyzed in each sample. TUBA is the loading control in immunoblots. Scale bars in confocal microscopy images represent 5 μm. All the presented microscopy and immunoblot data are representative of at least three independent experiments.](/cms/asset/e2e7f723-421a-42b6-b95d-c72472f3751b/kaup_a_1997053_f0005_oc.jpg)
A non-denaturing immunoprecipitation of endogenous LC3B from extract of MCF7 cells showed coimmunoprecipitation of HYPK with LC3B (). This observation was comparable with the findings of another recent study that also reported proximity-dependent HYPK interaction with ATG8/LC3 in plants [Citation49]. To validate if HYPK directly interacts with LC3 (MAP1LC3A/LC3A and MAP1LC3B/LC3B) and GABARAP (GABARAP, GABARAPL1 and GABARAPL2) class of proteins, we conducted protein-protein interaction analysis by affinity-isolation assays of recombinant proteins. We coexpressed 6xhistidine-tagged LC3A, LC3B, GABARAP, GABARAPL1, GABARAPL2 with untagged HYPK in BL21DE3 strain of Escherichia coli by using pETDuet-1 vector (pETDuet-1 vector allows coexpression of two recombinant proteins in T7 promoter expression system). Ni-NTA affinity-based isolation showed copurification of HYPK with all the LC3 and GABARAP proteins (), suggesting that HYPK could globally interact with LC3 and GABARAP subfamily proteins.
To probe the function of putative HYPK-LIR in LC3 binding, we used truncation mutations and alanine scanning of HYPK as described in . The HYPK-UBA was the C-terminal UBA domain, whereas HYPK-N84 construct was comprised of the N-terminal eighty-four amino acids (including the putative LIR). The HYPK Y49AEE52> A49AAA52 was a mutant construct in which 49th, 51st and 52nd amino acids (Y49, E51, and E52) were mutated to alanine (Y49A, E51A, and E52A) in the full-length HYPK. The binding assays of recombinant proteins by SPR showed that HYPK and HYPK-N84, but not the HYPK-UBA and HYPK Y49AEE52> A49AAA52, could bind to LC3B (), confirming that the LC3 binding site is in the N-terminal region of HYPK, and the LIR stretches from 49th to 52nd amino acid region. To validate the intracellular interaction of HYPK with LC3B, we traced the colocalization of HYPK, HYPK-N84, HYPK-UBA and HYPK Y49AEE52> A49AAA52 with LC3B protein in MCF7 cells. Similar to the observations of SPR study, only HYPK and HYPK-N84 showed high colocalization with LC3B (). Colocalization of HYPK with LC3B was also observed in HeLa and IMR-32 cells (Figure S6B). These results clearly show that HYPK interacts with LC3 by its atypical tyrosine-type (Y-type) LIR.
HYPK modulates autophagy
Having found that HYPK could interact with NEDD8 and LC3, we explored if HYPK modulated the intracellular induction and flux of autophagy. HYPK knockdown reduced the basal level of cellular autophagy, as seen by reduction of LC3B-II in HYPK-siRNA-treated MCF7, HeLa and IMR-32 cells compared to respective control cells (, ). HYPK knockdown also reduced the number of LC3B puncta, whereas HYPK overexpression increased the number of LC3B puncta in the above-mentioned cells (, 6D), indicating that HYPK helps in formation (i.e., induction) of autophagosomes. Imaging of HYPK overexpressing cells by transmission electron microscopy also showed increased autophagosome formation (), while the control cells contained significantly lesser number of autophagosomes ().
Figure 6. HYPK regulates initiation and flux of basal autophagy. (A) MCF7 cells were untransfected or transfected with control-siRNA or HYPK-siRNAs. Representative immunoblots of LC3B and HYPK from cell lysate. Densitometric quantification of LC3B-II and HYPK bands relative to TUBA of blots (* P < 0.05). (B) MCF7 cells were untransfected or transfected with HYPK-siRNA or HYPK overexpressing clone. Confocal immunofluorescence microscopy images of LC3B puncta representing potential autophagosomes in cells. Quantification (mean ± SD) of number of LC3B puncta in cells; ~ 200 cells analyzed in each sample (* P < 0.05). (C) Representative transmission electron micrographs of ultra-structures of IMR-32 cells. Cells in the – upper panel, left: untreated; upper panel, right: transfected with HYPK-overexpressing clone; lower panel, left: HTT97Q exon 1-expressing cells with endogenous expression of HYPK; lower panel, right: transfected with a plasmid encoding HYPK in HTT97Q exon 1. Quantitative (mean ± SD) estimation of autolysosomes in the above-mentioned cells, ~ 30 cells analyzed in each sample (* P < 0.05). (D) MCF7 cells were transfected with empty vector or HYPK overexpressing clones. Representative immunoblots of LC3B, BECN1, ATG5 and HYPK from cell lysate of time-chase experiments. Densitometric quantification of bands of above-mentioned proteins relative to TUBA of blots (* P < 0.05). (E) Stable RFP-GFP-LC3B expressing MCF7 cells were untransfected or transfected with control-siRNA or HYPK-siRNA. Confocal fluorescence microscopy images of GFP+ RFP+ LC3B autophagosomes and GFP− RFP+ LC3B autolysosomes. Quantitative count (mean ± SD) of GFP− RFP+ LC3B autolysosomes; ~ 100 cells analyzed in each sample (* P < 0.05). Immunoblot of HYPK from lysate of untreated, control-siRNA and HYPK-siRNA treated cells. TUBA is the loading control in immunoblots. Scale bars in confocal microscopy images: 5 μm. All the presented microscopy and immunoblot data are representative of at least three independent experiments.
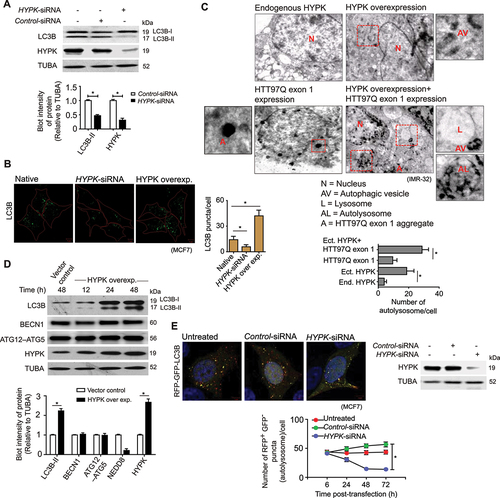
To further understand if HYPK was also involved in maintaining the autophagy flux, we conducted a time-chase experiment to monitor the cellular levels of LC3B-II, BECN1 and ATG12-ATG5 during overexpression of HYPK. Indeed, HYPK maintained the steady-state flux of autophagy by continuously stimulating the lipidation of LC3B-I to LC3B-II without changing the cellular level of BECN1 and ATG12–ATG5 (). Additionally, the maturation of autophagosomes to autolysosomes decreased in HYPK knockdown cells. The count of RFP+ GFP− LC3B puncta (ALs) were almost four-fold less in HYPK knockdown cells than control cells (). Thus, with these results, it was evident that HYPK functioned in selective autophagy induction and maturation.
HYPK facilitates neddylation-dependent aggrephagy during proteotoxic stress
To determine if HYPK clears the neddylated protein aggregates by autophagy during proteotoxic stress, we first analyzed the pattern of cellular distribution of HYPK and its colocalization with NEDD8 in puromycin-treated cells. We observed a time-dependent increase of HYPK foci, which colocalized with NEDD8 granules, in puromycin-treated MCF7 cells (, 7B). In fact, we had previously shown that self-oligomerization of HYPK sequestered heterogenous protein aggregates (such as, aggregates of SNCAA53T, SOD1G93A, HTT97Q exon 1, etc.) [Citation27]. Complementary results in this study showed that the formation of HYPK foci is dependent upon neddylation-status of protein aggregates in puromycin-treated cells. HYPK could not form self-oligomeric granules in NEDD8-KD cells compared to control cells (, 7D).
Figure 7. HYPK clears protein aggregates by polyneddylation-dependent aggrephagy. (A) MCF7 cells were treated with puromycin [15 μg/ml] for 0–18 h. Confocal immunofluorescence microscopy images of HYPK and NEDD8. Inset shows enlarged region of HYPK and NEDD8 colocalized granules marked with yellow rectangles. (B) Quantification of HYPK and NEDD8 positive granules in MCF7 cells with conditions as described in (a), ~ 100 cells analyzed in each sample. (C) MCF7 cells were untreated or treated with [15 μg/ml] puromycin for 24 h. Puromycin treatment was done in untransfected or control-siRNA or NEDD8-siRNA transfected cells. Confocal immunofluorescence microscopy images of HYPK. (D) Quantification (mean ± SD) of HYPK granules in MCF7 cells with conditions as described in (c), ~ 100 cells analyzed in each sample (* P < 0.05). (E and F) MCF7 cells were untransfected or transfected with control or HYPK-siRNA or ATG5-siRNA or PSMD8-siRNA. Cells were treated with [15 μg/ml] puromycin, followed by washout of puromycin as described in the timeline of the experiment. (E) confocal immunofluorescence microscopy images of NEDD8. (F) quantification of NEDD8 granules in cells with conditions as described in (E), ~ 100 cells analyzed in each sample (* P < 0.05). (G) Control-siRNA or HYPK-siRNA was transfected in NEDD8 overexpressing MCF7 cells. Left: representative immunoblots of LC3B, HYPK and NEDD8 from the lysates of untransfected or transfected cells. Right: Densitometric quantification of LC3B-II bands relative to TUBA of blots (* P < 0.05). (H) Confocal immunofluorescence microscopy images of HYPK, NEDD8, LC3B and LAMP1 in stable HTT97Q exon 1-GFP expressing IMR-32 cells. (I) Coefficient of colocalization of HYPK, LC3B and LAMP1 with neddylated HTT97Q exon 1-GFP, ~ 100 cells analyzed in each sample. (J) Immunoblot of HTT97Q exon 1 and HYPK from lysate of control and HYPK knockdown cells. Densitometric quantification of HTT97Q exon 1 bands relative to TUBA of blots (* P < 0.05). (K) 5 μM MG132 or 1 μM bafilomycin A1 was separately given for 24 h to stable HTT97Q exon 1 expressing IMR-32 cells that were untransfected or transfected with HYPK overexpressing clone. Representative immunoblots of HTT97Q exon 1 and HYPK. Densitometric quantification of HTT97Q exon 1 bands relative to TUBA of blots (* P < 0.05). (L) Control-siRNA or ATG5-siRNA was transfected in stable HTT97Q exon 1-expressing IMR-32 cells that also had HYPK overexpressing clone. Representative immunoblots of HTT97Q exon 1, ATG12–ATG5 and HYPK. Densitometric quantification of HTT97Q exon 1 bands relative to TUBA of blots (* P < 0.05). TUBA is the loading control in immunoblots. Scale bars in confocal microscopy images: 5 μm. All the presented microscopy and immunoblot data are representative of at least three independent experiments.
![Figure 7. HYPK clears protein aggregates by polyneddylation-dependent aggrephagy. (A) MCF7 cells were treated with puromycin [15 μg/ml] for 0–18 h. Confocal immunofluorescence microscopy images of HYPK and NEDD8. Inset shows enlarged region of HYPK and NEDD8 colocalized granules marked with yellow rectangles. (B) Quantification of HYPK and NEDD8 positive granules in MCF7 cells with conditions as described in (a), ~ 100 cells analyzed in each sample. (C) MCF7 cells were untreated or treated with [15 μg/ml] puromycin for 24 h. Puromycin treatment was done in untransfected or control-siRNA or NEDD8-siRNA transfected cells. Confocal immunofluorescence microscopy images of HYPK. (D) Quantification (mean ± SD) of HYPK granules in MCF7 cells with conditions as described in (c), ~ 100 cells analyzed in each sample (* P < 0.05). (E and F) MCF7 cells were untransfected or transfected with control or HYPK-siRNA or ATG5-siRNA or PSMD8-siRNA. Cells were treated with [15 μg/ml] puromycin, followed by washout of puromycin as described in the timeline of the experiment. (E) confocal immunofluorescence microscopy images of NEDD8. (F) quantification of NEDD8 granules in cells with conditions as described in (E), ~ 100 cells analyzed in each sample (* P < 0.05). (G) Control-siRNA or HYPK-siRNA was transfected in NEDD8 overexpressing MCF7 cells. Left: representative immunoblots of LC3B, HYPK and NEDD8 from the lysates of untransfected or transfected cells. Right: Densitometric quantification of LC3B-II bands relative to TUBA of blots (* P < 0.05). (H) Confocal immunofluorescence microscopy images of HYPK, NEDD8, LC3B and LAMP1 in stable HTT97Q exon 1-GFP expressing IMR-32 cells. (I) Coefficient of colocalization of HYPK, LC3B and LAMP1 with neddylated HTT97Q exon 1-GFP, ~ 100 cells analyzed in each sample. (J) Immunoblot of HTT97Q exon 1 and HYPK from lysate of control and HYPK knockdown cells. Densitometric quantification of HTT97Q exon 1 bands relative to TUBA of blots (* P < 0.05). (K) 5 μM MG132 or 1 μM bafilomycin A1 was separately given for 24 h to stable HTT97Q exon 1 expressing IMR-32 cells that were untransfected or transfected with HYPK overexpressing clone. Representative immunoblots of HTT97Q exon 1 and HYPK. Densitometric quantification of HTT97Q exon 1 bands relative to TUBA of blots (* P < 0.05). (L) Control-siRNA or ATG5-siRNA was transfected in stable HTT97Q exon 1-expressing IMR-32 cells that also had HYPK overexpressing clone. Representative immunoblots of HTT97Q exon 1, ATG12–ATG5 and HYPK. Densitometric quantification of HTT97Q exon 1 bands relative to TUBA of blots (* P < 0.05). TUBA is the loading control in immunoblots. Scale bars in confocal microscopy images: 5 μm. All the presented microscopy and immunoblot data are representative of at least three independent experiments.](/cms/asset/204a80a3-68f1-477b-881f-e4b02ae77ed0/kaup_a_1997053_f0007_oc.jpg)
To further investigate if HYPK has specific role in clearance of neddylated proteins through autophagy, we checked the formation and clearance of puromycin-induced neddylated granules in varying knockdown conditions of HYPK, ATG5 and PSMD8. Neddylated protein granules accumulated and persisted even after puromycin wash in HYPK-KD and ATG5-KD cells (, 7F). After puromycin wash, the load of neddylated granules in PSMD8-KD cells decreased to a minimum level that was comparable to control cells (, 7F). Additionally, HYPK-KD decreased the conversion of LC3B-I to LC3B-II, which was otherwise observed in NEDD8 overexpressing cells (). Such results implied that HYPK functions in the degradation of neddylated proteins through autophagy.
Having established the global role of HYPK in neddylation-dependent autophagy, we finally sought to elucidate if HYPK could channelize the degradation of neddylated HTT97Q exon 1 through aggrephagy. In IMR-32 cells, HYPK colocalized with NEDD8-positive HTT97Q exon 1-GFP aggregates (, 7I). LC3B and LAMP1 also colocalized with the protein complexes of HYPK and HTT97Q exon 1-GFP (, 7I). Increased fusion of autophagosomal HTT97Q exon 1 aggregates with lysosomes (forming autolysosomes) was observed in HYPK overexpressing cells (), suggesting that the complex of neddylated HTT97Q exon 1 aggregates were not only subjected to be enclosed into autophagic vacuoles, but they were also delivered to lysosomes for degradation. Besides measuring the steady-state degradation of HTT97Q exon 1 in HYPK knockdown condition (), HYPK’s role in autophagic degradation of neddylated HTT97Q exon 1 was confirmed by following the degradation of the later protein in different conditions that blocked the proteasomal or autophagy pathways. Although HYPK-facilitated degradation of HTT97Q exon 1 continued in presence of MG132 (), bafilomycin A1 treatment drastically prevented the capacity of HYPK to assist the degradation of HTT97Q exon 1 (). We further validated HYPK’s role as autophagy modulator of HTT97Q exon 1 in ATG5-KD/HYPK overexpressing cells. Clearance of HTT97Q exon 1 was impeded in those cells than control cells (). Based on these data, we conclude that autophagic degradation of neddylated proteins, including neddylated mutant HTT exon 1, plays crucial roles in active removal of toxic protein aggregates through the scaffolding functions of HYPK for NEDD8 and LC3 during proteotoxic stress.
Discussion
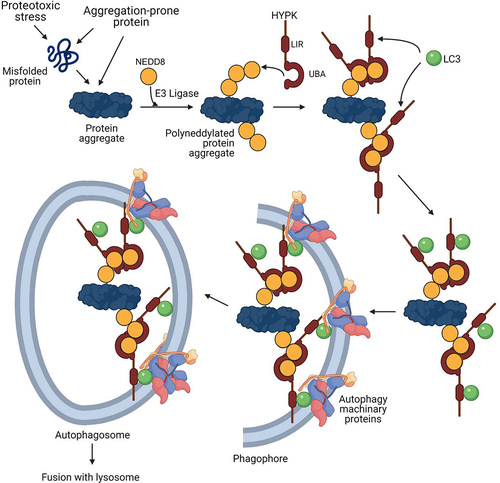
The various processes of clearance of intra- and extracellular protein aggregates are tailored to maintain proteostasis in misfolded protein disorders and neurodegenerative proteinopathies. The complexities in the mechanisms of autophagy are regulated by intriguing activity of different regulatory proteins. In this study, we show that NEDD8 and HYPK are stress-responsive regulators involved in degradation of protein aggregates by autophagy. The findings suggest that neddylation of cytosolic protein aggregates, and their inclusion in autophagosomes by the receptor function of HYPK () are parts of defensive response to control aggrephagy during proteotoxic stress.
Figure 8. Model of neddylation-dependent aggrephagy and the role of HYPK in this process. In conditions of intrinsic and/or extrinsic proteotoxic stress, intracellular protein aggregates are progressively neddylated. HYPK binds to NEDD8 of the polyneddylated protein aggregates by using its C-terminal UBA domain, followed by recruitment of LC3 to the site by HYPK’s LIR. This promotes autophagosomal enclosing of the neddylated protein aggregates for their subsequent degradation upon delivery to lysosome.
In the physiological condition, neddylation stabilizes the CUL protein of cullin-ring ligase (CRL) complexes [Citation50], mitochondrial proteins [Citation51] etc., other than regulating the activity of ubiquitin ligases [Citation52] and maintaining proper localization of ribosomal proteins [Citation53]. Polyneddylation of proteins leads to their degradation by proteasomal system. Here, we have shown that polyneddylation also prune the aggregated proteins for autophagic degradation in such a way that complements the protein ubiquitination function. We conceive that coexistence of polyubiquitination and polyneddylation pathways in aggrephagy is a part of fail-safe mechanism with far reaching consequences. The differences of ubiquitination and neddylation codes, as defined by the different kinds of lysine linkages in the chains, manifest alternative pathways of protein degradation. While K63-linked polyubiquitin chain delivers proteins to autophagosome, this position-specific lysine is absent in NEDD8. Thus, a unique, yet unknown, kind of neddylation linkage could generate additional topological selectivity for autophagy of proteins. Additionally, ubiquitination is a global process which is manifested in most of the cellular proteins, and a large fraction of polyubiquitinated proteins are intended for proteasomal degradation. Contrary to that, neddylation could only be occurring to a limited number of proteins in the direction of aggrephagy. Thus, superfluous neddylation of such proteins in stressed conditions could compromise their ubiquitination, thereby favoring proteins’ autophagic degradation.
Activation-dependent formation of homogeneous or mixed neddylation chains results in differential fate of the substrate proteins. Formation of polyneddylation chain consisting of only NEDD8 protein depends upon the initial charging of the NAE1 (of the NAE1-UBA3 heterodimeric E1 ligase complex) by activated NEDD8 [Citation54]. The observation that homogeneous polyneddylation of aggregation-prone proteins directs them to autophagic degradation supports the notion that unmixed chains of ubiquitin and NEDD8 are driving factors in protein degradation pathways, including autophagy. On the other hand, stress-induced formation of mixed ubiquitin-NEDD8 chain results due to UBA1-dependent incorporation of NEDD8 in polyubiquitin chain. Such ubiquitination system-induced NEDD8 addition to ubiquitin chain has an effect in further aggregation of proteins, although that phenomenon still participates in transient protection of cells from cytotoxicity [Citation20,Citation55].
HYPK regulates aggrephagy by interacting with LC3 and NEDD8, thereby defining the pivotal receptor function of this protein in a noncanonical autophagy. The charged and hydrophobic residues in the UBA domain are necessary for HYPK binding to NEDD8. The UBA domain is conserved in the longest isoform of HYPK protein of all species, signifying that NEDD8 binding is an essential function of HYPK. Although it is unknown what type of lysine-linkage in the polyneddylation chain specifies the autophagosomal degradation of proteins, it is reasonable to speculate that the unique sequence of HYPK-UBA domain is the determinant for recognizing such neddylation linkage. While HYPK recognizes the polyneddylation chain on the protein aggregates, we cannot exclude the fact that it also binds to monomeric NEDD8 and neddylation chain of soluble proteins.
The LC3 binding function is attributed to the N-terminal LIR of HYPK. HYPK has an unconventional LIR core sequence that lacks a hydrophobic amino acid at the fourth position. Although the LIR of HYPK can be categorized as a “composite LIR” [Citation56], more structural studies can expand the understanding if it also functions as “half LIR” [Citation57]. Nevertheless, our preliminary studies found that HYPK interacts with the LIR docking site (LDS) of LC3B, as LDS mutants of LC3B do not interact with HYPK (data not shown). The absence of hydrophobic interactions at the fourth position of HYPK-LIR could be compensated by an extended LIR that is inclusive of other hydrophobic interactions involving not only the HP1 region but also the HP0 and HP2 regions of LC3 and GABARAP proteins. Consistent with our previous report that the LIR-harboring N-terminal region of HYPK is a part of flexible and stretchable disordered nanostructure [Citation29], rearrangements of the essential interactions between HYPK-LIR and LDS are possible. While such rearrangements of interactions can lower the affinity of HYPK for LC3, oligomeric HYPK [Citation27] should bind multiple LC3 molecules, thereby increasing the overall binding events. Moreover, the LIR is present in all the isoforms of HYPK, including the splice variant (isoform 2 – NP_001186814.1) that lacks the UBA domain, suggesting that the LC3 binding and autophagy regulatory functions of HYPK are conserved.
The ability of HYPK to regulate neddylation-dependent autophagy links this protein to the general mechanisms of proteostasis. A workable model consists the proposition that proteotoxic stress causes the formation of protein aggregates which are successively neddylated. HYPK interacts with the NEDD8 of the neddylated protein aggregates, followed by sequestration of more HYPK with the aggregates due to HYPK’s cooperative self-association. Binding to NEDD8 possibly couples with HYPK’s transition from closed-to-open structure due to the loss of intramolecular interactions between the N-terminal charged region and C-terminal low complexity region. The open conformer of HYPK recruits LC3 to the aggresomes, followed by assembly of downstream autophagy proteins and autophagosomal enclosure of protein aggregates.
Aggrephagy is a major mode of alleviating the load of the pathological protein aggregates in cells of neurodegenerative disorders (NDs) [Citation58]. Our approach of a global screening of UBLs and UBA domain-containing proteins has identified and validated the functions of NEDD8 and HYPK in modulation of aggrephagy of mutant HTT exon 1 protein. NEDD8 and its conjugation machinery proteins have been identified as modulators of NDs-associated proteins [Citation59,Citation60]. Capturing of neddylated mutant HTT by NUB1 serves as the activation signal for proteasomal degradation of HTT protein. Besides the proteasomal pathway, our study shows that genetic and chemical perturbation of neddylation pathway inhibits the aggrephagy of insoluble mutant HTT exon 1 aggregates, thus, strongly suggesting that neddylation also recruits autophagosomal proteins to aggregates by generating NEDD8-HYPK-LC3 complex. The constitutive neddylation of mutant HTT could be a process of bipartite surveillance system for simultaneous and dispensable degradation of the protein by different pathways. The neddylation signal initially primes the soluble mutant HTT to proteasome, followed by autophagosomal removal of insoluble aggregates of the protein. It is reported that NUB1 expression, but not the NAE1 expression, is downregulated in knock-in mouse model of Huntington’s disease (HD) (GEO: GDS2912) [Citation61]. Therefore, it will be interesting to test whether autophagy is the primary mode of degradation of neddylated mutant HTT, although possibility of other supplementary mechanisms is also obvious. This information can also be extrapolated to speculate that neddylation is prerequisite for autophagic degradation of other labile proteins that are prone to misfolding and aggregation.
Similarly, the spatio-functional aspects of HYPK in proteostasis extend beyond its chaperone [Citation62] and aggregate-sequestering [Citation27] functions to nonredundant role in aggrephagy. Although HYPK reportedly exerts its chaperone activity by interacting with the N-terminal region of HTT [Citation63], we reveal that aggrephagy is the mechanism of HYPK-mediated removal of HTT aggregates. While the co-association of mutant HTT, HYPK, NEDD8 and LC3 could be dependent upon the context of layers of HTT aggregates, it is also interesting to note that the expression of HYPK is regulated by an array of other factors like thermal stress [Citation64], cellular aging [Citation33] etc. Our previous studies showed that not only HTT, but other disease-causing aggregation-prone proteins also elicit a HYPK coaggregation response [Citation27]. Furthermore, the recruitment of HYPK to neddylated proteins can also be remotely governed by the acetylation status of the target proteins. The interaction of HYPK with the proteins of N-acetyl transferase complex [Citation65] and acetylation-dependent neddylation of proteins [Citation66] are indicative of a complex crosstalk of post-translational modifications and HYPK recruitment in multiprotein complexes during aggrephagy of misfolded proteins. Overall, the neddylation-dependent aggrephagy takes place by synergistic effects of sequestration of HYPK with protein aggregates and tethering function of HYPK.
It is important to understand that HYPK expression is not significantly changed in different NDs and brain diseases (Table S1). Nevertheless, the potential translational value of neddylation-dependent autophagy and the role of HYPK in this process can be significant, considering that both NEDD8 and HYPK are positive modulators of aggrephagy. Critical changes of tissue- and organ-specific expression of NEDD8, neddylation machineries (such as ligases, deneddylation enzymes etc.) and HYPK by genetic or chemical processes can provide a unique mode for controlling the abundance of toxic protein aggregates which are otherwise accumulated in cells affected with NDs. Although NEDD8 and HYPK are not druggable targets due to their noncatalytic function, modulation of their expression could be a rewarding mean to cope with the challenges of protein aggregates. Thus, a chemical screening for identification of activators of NEDD8 and HYPK expression is warranted.
In summary, this study identified the functionality of NEDD8 and HYPK as the modulators of autophagy in context of clearance of proteotoxic stress-induced and innate protein aggregates, thereby describing a novel and noncanonical mechanism of aggrephagy that can be utilized in HD therapeutics.
Materials and methods
Clones and plasmids
To clone the open reading frames (ORFs) of different genes, total RNA was isolated (ThermoFischer Scientific [TFS], 12183018A) from IMR-32 cells, and cDNAs of corresponding mRNAs were made by reverse transcription using the oligo-dT primer (Eurofins Genomics [EG]). ORFs of genes were amplified from cDNAs by polymerase chain reaction (PCR) using specific primer sets (EG). The list of clones and plasmids used in this study is given in . The general process of cloning was similar to what was described in our previous studies [Citation27,Citation67]. Briefly, restriction digested PCR products and plasmids were ligated, followed by the transformation of the ligated products in ultra-competent DH5α strain of Escherichia coli. Positive clones were selected by colony PCR. All clones were sequenced at the sophisticated equipment facility of the research support service group of CDFD.
Table 1. Clones and plasmids.
Site-directed mutagenesis
Deletion mutants of HYPK were made by using specific primer sets (EG) in PCR-based method. FLAG or 6xhistine peptide sequence was introduced in different clones by incorporating the corresponding sequences in-frame at the N-terminus of ORFs using the forward primer (EG). Point mutations in HYPK (HYPK Y49AEE52> A49AAA52), UBA domain of HYPK (UBAD94A, E101A; UBA [ΔL113, ΔG118]) and NEDD8 (allR) were introduced by using multiple primer sets (EG) in overlapping PCR method. Mutations were confirmed by sequencing.
Recombinant protein production and purification
Recombinant HYPK, HYPK-UBA, HYPK-UBA D94A, E101A, HYPK-UBA [ΔL113, ΔG118], HYPK-N84, HYPK Y49AEE52> A49AAA52, NEDD8, LC3A, LC3B, GABARAP, GABARAPL1, GABARAPL2 proteins were produced by using a T7 expression system which was described by us in earlier studies [Citation29,Citation68]. The bacterial expression clones (in pET21 and pETDuet-1 vectors) were separately transformed in the BL21DE3 strain of Escherichia coli, followed by induction of protein synthesis by application of isopropyl β-D-1-thiogalactopyranoside (IPTG, 1 mM final concentration; Sigma Aldrich [SA], I6758) in the culture medium (Luria Bertani broth/ampicillin). After 12–18 h of protein production at 18°C, 180 rpm, bacterial cells were lysed by sonication in the lysis buffer (50 mM Tris-HCl, pH 8.0, 300 mM NaCl, 10 mM imidazole, 1 mM PMSF (SA, P7626). Cell lysate was cleared by centrifugation (14,000 g/40 min/4°C), followed by purification of proteins by nickel ion-based affinity exchange column chromatography. The cleared lysate was run through Ni+2-NTA agarose beads (Qiagen [QA], 30,250) to allow binding of the histidine-tagged (recombinant) proteins to beads. The beads were subsequently washed with wash buffer (50 mM Tris-HCl, pH 8.0, 300 mM NaCl, 40 mM imidazole), followed by elution of the proteins in elution buffer (50 mM Tris-HCl, pH 8.0, 300 mM NaCl, 300 mM imidazole). Proteins were dialyzed in different dialysis buffers depending upon the requirements of downstream experiments. In all the experiments, >98% pure proteins were used.
Protein-protein binding assay by surface plasmon resonance
The binding properties of HYPK, HYPK-UBA, HYPK-UBAD94A, E101A, HYPK-UBA [ΔL113, ΔG118] with NEDD8; and HYPK, HYPK-N84, HYPK-UBA, HYPK Y49AEE52> A49AAA52 with LC3B were analyzed by surface plasmon resonance technique. Using the NHS/EDC reagent, 5 nmole of NEDD8 or LC3B (in acetate buffer (pH 4.0)) was immobilized on a CM5 sensor chip (GE Healthcare Life Sciences, 29,149,604) by amine coupling. The mobile phase analytes (HYPK, HYPK-UBA, HYPK-UBA D94A, E101A and HYPK-UBA [ΔL113, ΔG118], HYPK-N84, HYPK Y49AEE52> A49AAA52) were kept in HBS-EP buffer (10 mM HEPES, pH 7.5, 150 mM NaCl, 3 mM EDTA, 0.005% [v:v] Surfactant P20 [Cytiva, BR100054; pH 7.4]). Analyte to ligand binding experiments were done in the Biacore 3000 instrument (GE Healthcare Life Sciences) at 25°C. Analytes were injected at a flowrate of 30 µl/min, and the dissociation events were allowed for 10 min. Concentrations of analytes were in the range of 40 nM-25 μM. Subtraction of nonspecific binding from the actual binding response was done by measuring the mock binding on the immobilized surface of another channel. Dissociation constants were generated by assuming 1:1 Langmuir model of interaction.
Cell culture and reagent treatment
The MCF7, HeLa, SH-SY5Y and IMR-32 cell-lines were procured from National Center for Cell Sciences (NCCS). MCF7 and HeLa cells were cultured in advanced DMEM medium (TFS, 12,491,023) which was supplemented with 10% fetal bovine serum (TFS, 26,140,095), 2 mM L-glutamine (TFS, 25,030,164) and 1x antibiotic-antimycotic solution (TFS, 11,548,876). IMR-32 and SH-SY5Y cells were grown in neuronal culture medium (TFS, 88,283) supplemented with other components as mentioned for MCF7 and HeLa cells. For most of the experiments, the choice of MCF7 as the model cell-line was based on the fact that this cell-line is recommended for many of the autophagy assays. Moreover, the autophagy proteins, NEDD8 and HYPK are adequately expressed in MCF7 cells, making it easy to follow the autophagy events during differential knock-down conditions and proteotoxic stress. Because IMR-32 is a neuroblastoma cell-line, it is more relevant to follow the autophagy events of neuronally expressed HTT (exon 1) in this cell-line. Cells were maintained in a humified incubator at 37°C with 5% CO2. Although cell-lines were not authenticated, they were routinely checked for mycoplasma contamination by using Venor™ GeM mycoplasma detection kit [SA, MP0025]. The stable expression of RFP-GFP-LC3B in MCF7 cell-line, and GFP-HTT97Q exon 1 or HTT97Q exon 1 in IMR-32 cell-line were selected by maintaining 2 μM neomycin (SA, PHR1491) in the cell culture medium.
Puromycin (SA, P8833), MLN4924 [SA (Calbiochem), 505,477), MG132 [SA (Calbiochem), 474,790], bafilomycin A1 (SA, 196,000), and MLN7243 (Aobious, AOB87172) were treated to cells with specific concentration for defined time (as mentioned in results).
Clones and plasmids were transiently transfected to subconfluent cells by using Lipofectamine 2000 (TFS, 11,668,019) and Opti-MEM (TFS, 31,985,070) medium following manufacturer’s instructions. siRNAs were transfected to cells by using Lipofectamine 3000 (TFS, L3000015). List of siRNAs are given in . Transfected cells were harvested at different timepoints after transfection depending upon the experimental requirements. In chase experiments, cells were intermittently harvested at an interval of 6 or 12 h.
Table 2. esiRNAs and siRNAs.
Fractionation of soluble proteins and insoluble protein aggregates from cell lysate
To separate the insoluble polyneddylated protein aggregates and/or HTT97Q exon 1 aggregates from the soluble proteins, cells were scrapped from cell culture dishes in phosphate-buffered saline (PBS, pH 7.4; TFS, 10,010,054), followed by lysis of cells in pre-chilled cell lysis buffer (LB – 50 mM Tris-HCl [pH 8.0], 50 mM NaCl, 5 mM MgCl2, 0.2% Triton X-100 [SA, T8787], 0.5% Nonidet P-40 [TFS, 85,142], 1x protease inhibitor cocktail [SA, 11,836,170,001], phosphatase inhibitor [SA, PHOSS-RO]). After keeping the cell suspension at 4°C for 15 min in LB, cell lysate was cleared by centrifugation at 14,000 g for 30 min (4°C). The supernatant contained the fraction of soluble proteins (SF). The precipitate, which contained insoluble fraction (IF) of protein aggregates and other cell debris, was resolubilized in LB that was supplemented with 1% SDS and 4 M guanidinium chloride, followed by centrifugation of the suspension at 14,000 g for 30 min (4°C). The supernatant, which contained high-molecular weight polyneddylated proteins and/or HTT97Q exon 1 protein, was finally dialyzed in dialysis buffer (DB: 50 mM Tris-HCl [pH 8.0], 50 mM NaCl).
Immunoprecipitation and immunoblotting
Immunoprecipitation (IP) of desired proteins were done from SF (2 mg of protein) or IF (1 mg of protein) by using crosslink magnetic IP/co-IP method (TFS, 88,805; Pierce Crosslink Magnetic IP/co-IP kit) following the manufacturer’s protocol. Some IP experiments were done in denaturing conditions (with 1% SDS) as mentioned in the result section. List of antibodies used in IPs is given in .
Table 3. Antibodies.
In immunoblotting (IB), 60–80 µg of total protein was added with 4x Laemmli buffer (Bio-Rad, 161–0747) and heated at 95°C for 10 min. Proteins were separated on 12% SDS-polyacrylamide gel, followed by transfer of proteins from gel to polyvinylidene fluoride (PVDF) membrane (Amersham Hybond P 0.45; GE Healthcare Life Sciences, 10,600,023) and sequential use of primary and secondary antibodies that are listed in . Intermittent washing was done by TBST buffer (SA, T9039). Chemiluminescent signals were generated by SuperSignal west femto maximum sensitivity substrate (TFS, 34,094) and detected by ChemiDoc XRS+ imaging system (Bio-Rad). Densitometric quantification of bands was done in ImageJ2 software [Citation69].
Immunocytochemistry and fluorescence microscopy
Cells were grown on glass coverslips. The adherent cells were washed with PBS, followed by fixation with methanol or 4% paraformaldehyde (in PBS). Cell permeabilization was done with 0.2% Triton X-100 (in PBS, pH 7.4), followed by blocking with 1% BSA (SA, A7906) in PBS. Primary and secondary antibodies (listed in ) were sequentially given to cells with intermittent washing with PBS. Cells were mounted in prolong antifade gold with or without DAPI (TFS, P36931; P10144). Images were acquired in LSM700 confocal laser scanning microscope (Carl Zeiss) with the 63x Plan-Apochromat/1.4 NA oil/DIC M27 objective. Images were processed in Zen-Lite 2010 software (Carl Zeiss). Colocalization analysis of different proteins was done in Zen-Lite 2010 (blue edition) software (Carl Zeiss) by using the colocalization algorithm. The Pearson’s correlation coefficient was used as a measure of colocalization of proteins.
The LC3B-positive autophagosome puncta, NEDD8 granules and HTT97Q exon 1-GFP aggregates in cells were counted in Eclips Ti E (Nikon) microscope by using NIS-elements imaging software.
Autophagy flux assay
The GFP+ RFP+ and GFP− RFP+ LC3B puncta represented the autophagosomes and autolysosomes respectively, and their numbers were used to measure the autophagy flux as mentioned in previous study [Citation70]. To monitor the autophagy flux in varying knockdown condition of different proteins, cells were transfected with 10 nM of esiRNA against target mRNA. The culture medium was changed after 48 h of transfection.
Transmission electron microscopy
IMR-32 cells were grown in culture dishes. HYPK, HTT97Q exon 1 or HTT97Q exon 1+ HYPK were transfected to cells by the procedure that is mentioned in cell culture section. Cells were detached from dishes by trypsin-EDTA solution (SA, T3924). Detached cells were fixed in 1.5% (v:v) glutaraldehyde/4% (w:v) formaldehyde (in 0.1 M cacodylate buffer, pH 7.4) solution for five hours, followed by washing of cells with PBS (pH 7.4) for few times. Dehydration of the cells was gradually done by sequentially keeping them in 50%, 70% and 90% ethanol (15 min/solution). Cells were sectioned (1 μm thick) in ultra-microtome before placing them on 200 mesh copper grid (SA, G4776). Cell-section containing grids were incubated in 0.05 M glycine (in PBS) for 20 min. Grids were then washed with PBS, followed by staining with aqueous uranyl acetate for 2 min. After the grids were washed with water, they were transiently (for 15 s) exposed to lead citrate. Images were collected in Tecnai G2 Spirit Bio-TWIN transmission electron microscope. Electron beam strength was 15 kV and images were recorded by Gatan Orins CCD camera.
Computational studies
Protein-protein docking
Structures of NEDD8 protein and the UBA domains of different human proteins were curated (listed in ) from the Protein Data Bank [Citation71]. Structures of the proteins were optimized to add missing hydrogen atoms and assign bond orders in the protein preparation wizard of Maestro 9.2 (Schrödinger) [forcefield: OPLS_2005, convergence of heavy atoms to root mean square deviation (RMSD): 0.3 Å] as described in our previous studies [Citation72,Citation73]. Structures were subsequently minimized in molecular modeling toolkit [Citation74] by assisted model building with energy refinement (AMBER) [Citation75] simulation to 103 steepest descent iteration. Unrestrained docking of the finally prepared protein structures was done by using the PIPER program [Citation76] in the BioLuminate suite (Maestro 9.2, Schrödinger). The stabilities of complexes were measured by analyzing the ΔG(diss) in the PDBePISA (Proteins, Interfaces, Structures and Assemblies) web server [Citation77]. Structures were viewed and interpreted in PyMOL.
Table 4. PDB identifiers of protein structures.
Sequence alignment
The sequences of ubiquitin, UBLs and HYPK of different organisms were obtained from protein database of National Center for Biotechnology Information (NCBI). Sequences of UBA domains of different human proteins were curated from the SMART database [Citation93]. The alignments of sequences were done in Clustal Omega [Citation94].
Phylogenetic analysis
The sequence alignment file of the UBA domains of different human proteins was saved in the phylip (.phy) format which was further used in the Phylip-3.695 software package. The Seqboot, Protdist, Neighbor and Consense programs were sequentially run to analyze the phylogeny of the UBA domains. The outfile of each program was taken as the input file of the next program. Details of the parameters of each program are available upon request. The cladogram was generated by the Treeview software.
Statistical analysis
The statistical significance of difference of means between groups were analyzed by two-tailed, homoscedastic student’s t-test. P < 0.05 represented statistically significant difference.
Graphics
The graphics were made in Adobe Illustrator.
Supplemental Material
Download MS Word (3.6 MB)Acknowledgments
The authors extend their thankfulness to the Biocluster of National Centre for Biological Sciences (India) for providing the TEM facility. The authors also thank the members of CFG group for giving critical inputs in different experiments and manuscript writing. Authors acknowledge Mrs. Sreya Dey Ghosh for proofreading the manuscript. The personnel of sophisticated equipment facility of the research support service group of CDFD are acknowledged for the technical assistance in operating different instruments.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Supplementary material
Supplemental data for this article can be accessed here
Additional information
Funding
References
- Labbadia J, Morimoto RI. The biology of proteostasis in aging and disease. Annu Rev Biochem. 2015;84(1):435–464.
- Klaips CL, Jayaraj GG, Hartl FU. Pathways of cellular proteostasis in aging and disease. J Cell Biol. 2018;217(1):51–63.
- Chen B, Retzlaff M, Roos T, et al. Cellular strategies of protein quality control. Cold Spring Harb Perspect Biol. 2011;3:a004374.
- Kim YE, Hipp MS, Bracher A, et al. Molecular chaperone functions in protein folding and proteostasis. Annu Rev Biochem. 2013;82(1):323–355.
- Tanaka K. The proteasome: overview of structure and functions. Proc Jpn Acad Ser B Phys Biol Sci. 2009;85(1):12–36.
- Glick D, Barth S, Macleod KF. Autophagy: cellular and molecular mechanisms. J Pathol. 2010;221(1):3–12.
- Lamark T, Johansen T. Aggrephagy: selective disposal of protein aggregates by macroautophagy. Int J Cell Biol. 2012;(2012:736905.
- Mizushima N. Autophagy: process and function. Genes Dev. 2007;21(22):2861–2873.
- Hurley JH, Young LN. Mechanisms of autophagy initiation. Annu Rev Biochem. 2017;86(1):225–244.
- Wesch N, Kirkin V, Rogov VV. Atg8-family proteins-structural features and molecular interactions in autophagy and beyond. Cells. 2020;9(9):2008.
- Ruz C, Alcantud JL, Vives Montero F, et al. Proteotoxicity and neurodegenerative diseases. Int J Mol Sci. 2020;21(16):5646.
- Chen RH, Chen YH, Huang TY. Ubiquitin-mediated regulation of autophagy. J Biomed Sci. 2019;26:80.
- Zheng N, Shabek N. Ubiquitin ligases: structure, function, and regulation. Annu Rev Biochem. 2017;86(1):129–157.
- Tan JM, Wong ESP, Kirkpatrick DS, et al. Lysine 63-linked ubiquitination promotes the formation and autophagic clearance of protein inclusions associated with neurodegenerative diseases. Hum Mol Genet. 2008;17(3):431–439.
- Lorente M, et al. Inhibiting SUMO1-mediated SUMOylation induces autophagy-mediated cancer cell death and reduces tumour cell invasion via RAC1. J Cell Sci. 2019;132. DOI:https://doi.org/10.1242/jcs.234120.
- Enchev RI, Schulman BA, Peter M. Protein neddylation: beyond cullin-RING ligases. Nat Rev Mol Cell Biol. 2015;16:30–44.
- Abidi N, Xirodimas DP. Regulation of cancer-related pathways by protein NEDDylation and strategies for the use of NEDD8 inhibitors in the clinic. Endocr Relat Cancer. 2015;22:T55–70.
- Jayabalan AK, Sanchez A, Park RY, et al. NEDDylation promotes stress granule assembly. Nat Commun. 2016;7(1):12125.
- Mori F, Nishie M, Piao Y-S, et al. Accumulation of NEDD8 in neuronal and glial inclusions of neurodegenerative disorders. Neuropathol Appl Neurobiol. 2005;31(1):53–61.
- Maghames CM, Lobato-Gil S, Perrin A, et al. NEDDylation promotes nuclear protein aggregation and protects the Ubiquitin Proteasome System upon proteotoxic stress. Nat Commun. 2018;9(1):4376.
- Lane DP. Stress, specificity and the NEDD8 proteome. Cell Cycle. 2012;11(8):1488–1489.
- Li J, Ma W, Li H, et al. NEDD8 Ultimate Buster 1 Long (NUB1L) protein suppresses atypical neddylation and promotes the proteasomal degradation of misfolded proteins. J Biol Chem. 2015;290(39):23850–23862.
- Behrends C, Fulda S. Receptor proteins in selective autophagy. Int J Cell Biol. 2012;2012:673290.
- Lippai M, Low P. The role of the selective adaptor p62 and ubiquitin-like proteins in autophagy. Biomed Res Int. 2014;2014:832704.
- Sarraf SA. Loss of TAX1BP1-directed autophagy results in protein aggregate accumulation in the brain. Mol Cell. 2020;80:779–795. e710.
- Lu K, Psakhye I, Jentsch S, et al. Autophagic clearance of polyQ proteins mediated by ubiquitin-Atg8 adaptors of the conserved CUET protein family. Cell. 2014;158(3):549–563.
- Ghosh DK, Roy A, Aggregation-prone RA, et al. Regions in HYPK help it to form sequestration complex for toxic protein aggregates. J Mol Biol. 2018;430:963–986.
- Ghosh DK, Ranjan A. An IRES-dependent translation of HYPK mRNA generates a truncated isoform of the protein that lacks the nuclear localization and functional ability. RNA Biol. 2019;16:1604–1621.
- Ghosh DK, Roy A, Ranjan A, et al. Disordered nanostructure in huntingtin interacting protein K acts as a stabilizing switch to prevent protein aggregation. Biochemistry. 2018;57(13):2009–2023.
- Ghosh DK, Kumar A, Ranjan A, et al. Metastable states of HYPK-UBA domain’s seeds drive the dynamics of its own aggregation. Biochim Biophys Acta Gen Subj. 2018;1862:2846–2861.
- Arnesen T, Starheim KK, Van Damme P, et al. The chaperone-like protein HYPK acts together with NatA in cotranslational N-terminal acetylation and prevention of Huntingtin aggregation. Mol Cell Biol. 2010;30(8):1898–1909.
- Ayyadevara S, Balasubramaniam M, Gao Y, et al. Proteins in aggregates functionally impact multiple neurodegenerative disease models by forming proteasome-blocking complexes. Aging Cell. 2015;14(1):35–48.
- Bell R, Hubbard A, Chettier R, et al. A human protein interaction network shows conservation of aging processes between human and invertebrate species. PLoS Genet. 2009;5(3):e1000414.
- Xu G. Vulnerability of newly synthesized proteins to proteostasis stress. J Cell Sci. 2016;129:1892–1901.
- Azzam ME, Algranati ID. Mechanism of puromycin action: fate of ribosomes after release of nascent protein chains from polysomes. Proc Natl Acad Sci U S A. 1973;70(12):3866–3869.
- Yewdell JW, Anton LC, Bennink JR, et al. Defective ribosomal products (DRiPs): a major source of antigenic peptides for MHC class I molecules? J Immunol. 1996;157:1823–1826.
- Wenger T, Terawaki S, Camosseto V, et al. Autophagy inhibition promotes defective neosynthesized proteins storage in ALIS, and induces redirection toward proteasome processing and MHCI-restricted presentation. Autophagy. 2012;8(3):350–363.
- Ciuffa R, Lamark T, Tarafder A, et al. The selective autophagy receptor p62 forms a flexible filamentous helical scaffold. Cell Rep. 2015;11(5):748–758.
- Russell RC, Tian Y, Yuan H, et al. ULK1 induces autophagy by phosphorylating Beclin-1 and activating VPS34 lipid kinase. Nat Cell Biol. 2013;15(7):741–750.
- Lu B, Al-Ramahi I, Valencia A, et al. Identification of NUB1 as a suppressor of mutant Huntington toxicity via enhanced protein clearance. Nat Neurosci. 2013;16(5):562–570.
- Schulman BA, Harper JW. Ubiquitin-like protein activation by E1 enzymes: the apex for downstream signalling pathways. Nat Rev Mol Cell Biol. 2009;10(5):319–331.
- Leidecker O, Matic I, Mahata B, et al. The ubiquitin E1 enzyme Ube1 mediates NEDD8 activation under diverse stress conditions. Cell Cycle. 2012;11(6):1142–1150.
- Liu S, Yang H, Zhao J, et al. NEDD8 ultimate buster-1 long (NUB1L) protein promotes transfer of NEDD8 to proteasome for degradation through the P97UFD1/NPL4 complex. J Biol Chem. 2013;288(43):31339–31349.
- Nedelsky NB, Todd PK, Taylor JP, et al. Autophagy and the ubiquitin-proteasome system: collaborators in neuroprotection. Biochim Biophys Acta. 2008;1782:691–699.
- Su V, Lau AF. Ubiquitin-like and ubiquitin-associated domain proteins: significance in proteasomal degradation. Cell Mol Life Sci. 2009;66:2819–2833.
- Tanaka T, Kawashima H, Yeh ET, et al. Regulation of the NEDD8 conjugation system by a splicing variant, NUB1L. J Biol Chem. 2003;278:32905–32913.
- Gottlieb L., Marmorstein R. Structure of human NatA and its regulation by the huntingtin interacting protein HYPK. Structure. 2018;26(7):925–935. e928.
- Habisov S, Huber J, Ichimura Y, et al. Structural and functional analysis of a novel interaction Motif within UFM1-activating Enzyme 5 (UBA5) required for binding to Ubiquitin-like proteins and Ufmylation. J Biol Chem. 2016;291(17):9025–9041.
- Macharia MW, Tan WYZ, Das PP, et al. Proximity-dependent biotinylation screening identifies NbHYPK as a novel interacting partner of ATG8 in plants. BMC Plant Biol. 2019;19:326.
- Keuss MJ, Hjerpe R, Hsia O, et al. Unanchored tri-NEDD8 inhibits PARP-1 to protect from oxidative stress-induced cell death. EMBO J. 2019;38(6). DOI:https://doi.org/10.15252/embj.2018100024
- Zhang X, Zhang Y-L, Qiu G, et al. Hepatic neddylation targets and stabilizes electron transfer flavoproteins to facilitate fatty acid beta-oxidation. Proc Natl Acad Sci U S A. 2020;117(5):2473–2483.
- Xie P, Zhang M, He S, et al. The covalent modifier Nedd8 is critical for the activation of Smurf1 ubiquitin ligase in tumorigenesis. Nat Commun. 2014;5(1):3733.
- Sundqvist A, Liu G, Mirsaliotis A, et al. Regulation of nucleolar signalling to p53 through NEDDylation of L11. EMBO reports. 2009;10(10):1132–1139.
- Gong L, Yeh ET. Identification of the activating and conjugating enzymes of the NEDD8 conjugation pathway. J Biol Chem. 1999;274:12036–12042.
- Lobato-Gil S, Heidelberger JB, Maghames C, et al. Proteome-wide identification of NEDD8 modification sites reveals distinct proteomes for canonical and atypical NEDDylation. Cell Rep. 2021;34(3):108635.
- Johansen T, Lamark T. Selective autophagy: ATG8 family proteins, LIR motifs and cargo receptors. J Mol Biol. 2020;432(1):80–103.
- Mandell MA, Kimura T, Jain A, et al. TRIM proteins regulate autophagy: TRIM5 is a selective autophagy receptor mediating HIV-1 restriction. Autophagy. 2014;10(12):2387–2388.
- Yamamoto A, Simonsen A. The elimination of accumulated and aggregated proteins: a role for aggrephagy in neurodegeneration. Neurobiol Dis. 2011;43(1):17–28.
- Richet E, Pooler AM, Rodriguez T, et al. NUB1 modulation of GSK3beta reduces tau aggregation. Hum Mol Genet. 2012;21(24):5254–5267.
- Tanji K, Mori F, Kakita A, et al. Immunohistochemical localization of NUB1, a synphilin-1-binding protein, in neurodegenerative disorders. Acta Neuropathol. 2007;114(4):365–371.
- Hodges A, Hughes G, Brooks S, et al. Brain gene expression correlates with changes in behavior in the R6/1 mouse model of Huntington’s disease. Genes Brain Behav. 2008;7(3):288–299.
- Raychaudhuri S, Sinha M, Mukhopadhyay D, et al. HYPK, a Huntingtin interacting protein, reduces aggregates and apoptosis induced by N-terminal Huntingtin with 40 glutamines in Neuro2a cells and exhibits chaperone-like activity. Hum Mol Genet. 2008;17(2):240–255.
- Choudhury KR, Bhattacharyya NP. Chaperone protein HYPK interacts with the first 17 amino acid region of Huntingtin and modulates mutant HTT-mediated aggregation and cytotoxicity. Biochem Biophys Res Commun. 2015;456(1):66–73.
- Sakurai H, Sawai M, Ishikawa Y, et al. Heat shock transcription factor HSF1 regulates the expression of the Huntingtin-interacting protein HYPK. Biochim Biophys Acta. 2014;1840(3):1181–1187.
- Weyer FA. Structural basis of HypK regulating N-terminal acetylation by the NatA complex. Nat Commun. 2017;8:15726.
- Scott DC. Blocking an N-terminal acetylation-dependent protein interaction inhibits an E3 ligase. Nat Chem Biol. 2017;13:850–857.
- Ghosh DK, Roy A, Ranjan A. The ATPase VCP/p97 functions as a disaggregase against toxic Huntingtin exon 1 aggregates. FEBS Lett. 2018;592:2680–2692.
- Kumar A, Ghosh DK, Ali J, et al. Characterization of lipid binding properties of plasmodium falciparum Acyl-Coenzyme a binding proteins and their competitive inhibition by mefloquine. ACS Chem Biol. 2019;14:901–915.
- Rueden CT, Schindelin J, Hiner MC, et al. ImageJ2: imageJ for the next generation of scientific image data. BMC Bioinformatics. 2017;18(1):529.
- Loos B, du Toit A, Hofmeyr JH. Defining and measuring autophagosome flux-concept and reality. Autophagy. 2014;10:2087–2096.
- Berman HM, et al. The protein data bank. Nucleic Acids Res. 2000;28(1):235–242.
- Ghosh DK, Shrikondawar AN, Ranjan A. Local structural unfolding at the edge-strands of beta sheets is the molecular basis for instability and aggregation of G85R and G93A mutants of Superoxide dismutase 1. J Biomol Struct Dyn. 2019;38(3):647–659.
- Ghosh DK, Kumar A, Ranjan A. T54R mutation destabilizes the dimer of Superoxide dismutase T54R by inducing steric clashes at the dimer interface. RSC Adv. 2020;10:10776–10788.
- Hinsen K. The molecular modeling toolkit: a new approach to molecular simulations. J Comput Chem. 2000;21(2):79–85.
- Case DA, Cheatham TE, Darden T, et al. The Amber biomolecular simulation programs. J Comput Chem. 2005;26(16):1668–1688.
- Kozakov D, Brenke R, Comeau SR, et al. PIPER: an FFT-based protein docking program with pairwise potentials. Proteins. 2006;65:392–406.
- Krissinel E, Henrick K. Inference of macromolecular assemblies from crystalline state. J Mol Biol. 2007;372:774–797.
- Kozlov G, Peschard P, Zimmerman B, et al. Structural basis for UBA-mediated dimerization of c-Cbl ubiquitin ligase. J Biol Chem. 2007;282(37):27547–27555.
- Zhou ZR, Gao H-C, Zhou C-J, et al. Differential ubiquitin binding of the UBA domains from human c-Cbl and Cbl-b: NMR structural and biochemical insights. Protein Sci. 2008;17(10):1805–1814.
- Sander B, Xu W, Eilers M, et al. A conformational switch regulates the ubiquitin ligase HUWE1. Elife. 2017;6. DOI:https://doi.org/10.7554/eLife.21036.
- Marx A. Structural variations in the catalytic and ubiquitin-associated domains of microtubule-associated protein/microtubule affinity regulating kinase (MARK) 1 and MARK2. J Biol Chem. 2006;281:27586–27599.
- Panneerselvam S, Marx A, Mandelkow EM, et al. Structure of the catalytic and ubiquitin-associated domains of the protein kinase MARK/Par-1. Structure. 2006;14:173–183.
- Murphy JM. Conformational instability of the MARK3 UBA domain compromises ubiquitin recognition and promotes interaction with the adjacent kinase domain. Proc Natl Acad Sci U S A. 2007;104:14336–14341.
- Walinda E, Morimoto D, Sugase K, et al. Solution structure of the ubiquitin-associated (UBA) domain of human autophagy receptor NBR1 and its interaction with ubiquitin and polyubiquitin. J Biol Chem. 2014;289(20):13890–13902.
- Whitby FG, Xia G, Pickart CM, et al. Crystal structure of the human ubiquitin-like protein NEDD8 and interactions with ubiquitin pathway enzymes. J Biol Chem. 1998;273(52):34983–34991.
- Withers-Ward ES, Mueller TD, Chen IS, et al. Biochemical and structural analysis of the interaction between the UBA(2) domain of the DNA repair protein HHR23A and HIV-1 Vpr. Biochemistry. 2000;39:14103–14112.
- Ryu KS, Lee K-J, Bae S-H, et al. Binding surface mapping of intra- and interdomain interactions among hHR23B, ubiquitin, and polyubiquitin binding site 2 of S5a. J Biol Chem. 2003;278(38):36621–36627.
- Evans CL, Long JE, Gallagher TR, et al. Conformation and dynamics of the three-helix bundle UBA domain of p62 from experiment and simulation. Proteins. 2008;71:227–240.
- Middleton AJ, Day CL. The molecular basis of lysine 48 ubiquitin chain synthesis by Ube2K. Sci Rep. 2015;5(1):16793.
- Zhang D, Raasi S, Fushman D, et al. Affinity makes the difference: nonselective interaction of the UBA domain of Ubiquilin-1 with monomeric ubiquitin and polyubiquitin chains. J Mol Biol. 2008;377:162–180.
- Kieken F, Spagnol G, Su V, et al. NMR structure note: UBA domain of CIP75. J Biomol NMR. 2010;46:245–250.
- Chang YG, Song A-X, Gao Y-G, et al. Solution structure of the ubiquitin-associated domain of human BMSC-UbP and its complex with ubiquitin. Protein Sci. 2006;15(6):1248–1259.
- Ponting CP, Schultz J, Milpetz F, et al. SMART: identification and annotation of domains from signalling and extracellular protein sequences. Nucleic Acids Res. 1999;27(1):229–232.
- Sievers F. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol. 2011;7:539.
- Lesnick TG. A genomic pathway approach to a complex disease: axon guidance and Parkinson disease. PLoS Genet. 2007;3(6):e98.
- Dunckley T. Gene expression correlates of neurofibrillary tangles in Alzheimer’s disease. Neurobiol Aging. 2006;27:1359–1371.
- Lee JM, Zhang J, Su AI, et al. A novel approach to investigate tissue-specific trinucleotide repeat instability. BMC Syst Biol. 2010;4(1):29.
- Ferraiuolo L, Heath PR, Holden H, et al. Microarray analysis of the cellular pathways involved in the adaptation to and progression of motor neuron injury in the SOD1 G93A mouse model of familial ALS. J Neurosci. 2007;27(34):9201–9219.
- Chen-Plotkin AS, Geser F, Plotkin JB, et al. Variations in the progranulin gene affect global gene expression in frontotemporal lobar degeneration. Hum Mol Genet. 2008;17(10):1349–1362.
- Aydin D, Filippov MA, Tschäpe J-A, et al. Comparative transcriptome profiling of amyloid precursor protein family members in the adult cortex. BMC Genomics. 2011;12(1):160.
- Gatchel JR, Watase K, Thaller C, et al. The insulin-like growth factor pathway is altered in spinocerebellar ataxia type 1 and type 7. Proc Natl Acad Sci U S A. 2008;105(4):1291–1296.
- Han MH, Lundgren DH, Jaiswal S, et al. Janus-like opposing roles of CD47 in autoimmune brain inflammation in humans and mice. J Exp Med. 2012;209(7):1325–1334.
- Lockstone HE, Harris LW, Swatton JE, et al. Gene expression profiling in the adult Down syndrome brain. Genomics. 2007;90(6):647–660.
- Kim MY, Shu Y, Carsillo T, et al. hsp70 and a novel axis of type I interferon-dependent antiviral immunity in the measles virus-infected brain. J Virol. 2013;87(2):998–1009.
- Gersten M, Alirezaei M, Marcondes MCG, et al. An integrated systems analysis implicates EGR1 downregulation in simian immunodeficiency virus encephalitis-induced neural dysfunction. J Neurosci. 2009;29(40):12467–12476.
- Hur J, Sullivan KA, Pande M, et al. The identification of gene expression profiles associated with progression of human diabetic neuropathy. Brain. 2011;134(11):3222–3235.