
ABSTRACT
Mitochondria are critical organelles that maintain cellular metabolism and overall function. The catabolic pathway of autophagy plays a central role in recycling damaged mitochondria. Although the autophagy pathway is indispensable for some cancer cell survival, our latest study shows that rare autophagy-dependent cancer cells can adapt to loss of this core pathway. In the process, the autophagy-deficient cells acquire unique dependencies on alternate forms of mitochondrial homeostasis. These rare autophagy-deficient clones circumvent the lack of canonical autophagy by increasing mitochondrial dynamics and by recycling damaged mitochondria via mitochondrial-derived vesicles (MDVs). These studies are the first to implicate MDVs in cancer cell metabolism although many unanswered questions remain about this non-canonical pathway.
Mitochondria are highly organized and dynamic organelles that play fundamental roles in cellular metabolism, signaling, and viability. Cells maintain mitochondrial homeostasis through a conserved, genetically programmed, catabolic process known as mitophagy. Mitophagy starts with the formation of a double-membrane structure called a phagophore that engulfs damaged mitochondria. Subsequently, the phagophore matures into an autophagosome (also referred to as a mitophagosome during mitophagy), which then fuses with a lysosome leading to mitochondrial degradation. Impaired mitophagy contributes to the pathogenesis of several human diseases including cancer and neurodegeneration. Using a quantitative live-cell imaging CRISPR assay, we showed some cancer cell lines are especially dependent on the mitophagy/ autophagy pathway for survival [Citation1]. However, while knockout of core autophagy genes such as ATG7, or RB1CC1/FIP200, causes most of the cells to die, rare cells within the autophagy-dependent population can escape loss of this core pathway. We discovered that as the cells adapt to autophagy inhibition, they acquire new dependencies to maintain mitochondrial homeostasis including altered mitochondrial dynamics and increased mitochondrial-derived vesicles ().
Figure 1. Inhibition of the canonical mitophagy pathway in autophagy-dependent cancer cells forces rare surviving clones to undergo adaptation. The surviving autophagy-deficient cells upregulate mitochondrial fusion and generate mitochondrial-derived vesicles as an alternative means to recycle damaged mitochondria and maintain cellular homeostasis.
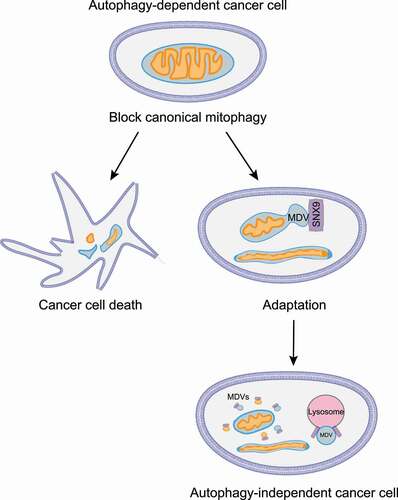
The autophagy-deficient cancer cells generated from originally autophagy-dependent cells are also deficient for canonical mitophagy, but they can still respond to mitochondrial-targeted insults and maintain normal mitochondrial function. We show that these rare autophagy-deficient cells use mitochondrial fusion as an adaptive mechanism to maintain cell function. The autophagy-deficient cells have increased expression of the dynamin-related GTPases MFN1 (mitofusin 1) and MFN2 that mediate outer-mitochondrial membrane (OMM) fusion without a change in total mitochondrial mass. Time-lapse electron-microscopy reveals an increase in active mitochondrial fusion events in the adapted autophagy-deficient cells leading to a more hyperfused phenotype compared to WT cells. Additionally, inhibition of mitochondrial fusion with a dominant-negative MFN1 protein significantly decreases growth and increases apoptosis in autophagy-deficient cell populations. Together, these data suggest autophagy-deficient cells derived from autophagy-dependent cells gain an acquired dependency on mitochondrial fusion to maintain mitochondrial function and cell survival.
Active mitochondrial fusion is sufficient to compensate for loss of mitophagy under basal conditions. However, increased mitochondrial fusion does not explain how autophagy-deficient cells cope with extreme mitochondrial damage. Instead, we discovered that the autophagy-deficient cells utilize a second alternative system for mitochondrial degradation and recycling – mitochondrial-derived vesicles (MDVs). MDVs are ~70- to 150-nm (apparent size ~500 nm due to limited resolution of confocal microscope) vesicles generated from the inner or outer mitochondrial membrane and preferentially contain proteins from these respective compartments. While these structures are highly understudied, others have used electron microscopy to show that outer-membrane derived MDVs are single-membrane vesicles while inner-membrane derived MDVs contain 2 membranes but still exclude outer-membrane proteins. These vesicles deliver pieces of damaged mitochondria directly into lysosomes, therefore completely bypassing the canonical autophagosome-mediated lysosomal degradation pathway. Using high-resolution confocal microscopy, we show that autophagy-deficient cells form more MDVs that traffic to lysosomes. Additionally, treatment with mitochondrial-targeting drugs such as the iron chelator DFP, or the potent uncoupler of mitochondrial oxidative phosphorylation CCCP, significantly increases MDV production in both WT cells and to an even greater extent in autophagy-deficient cells. MDV flux through lysosomes was quantitatively measured by ratiometric flow cytometry to quantify pH-sensitive probes including mCherry-GFP-FIS1 (better known as mito-QC). Flow cytometry with the FIS1-tandem probe shows that autophagy-deficient cells increase autophagosome-independent flux of MDVs through lysosomes. Furthermore, we showed that autophagy-deficient cells develop an acquired dependency on the endocytic protein SNX9 (sorting nexin 9) for survival. SNX9 is necessary for the formation of MDVs on mitochondria and their delivery into lysosomes. shRNA-mediated knockdown of SNX9 significantly reduces mitochondrial damage-induced MDV production in autophagy-deficient cells. Ratiometric flow cytometry also reveals that SNX9 acts similar to other autophagy adapter proteins and gets degraded itself during the process of trafficking MDVs to lysosomes. Thus, SNX9-mediated MDVs are an alternate form of mitochondrial homeostasis that compensate for loss of canonical autophagosome-dependent mitophagy and maintain overall mitochondrial health.
While SNX9 is important for MDV production and lysosomal delivery, we speculate that a plethora of additional genes are required in the MDV pathway. There are approximately 1,000 known mitochondrial proteins and it is still unclear which of these proteins might facilitate MDV-related mitochondrial membrane curvature, coat complex formation, the final scission event, lysosomal delivery, and fusion, among others. A more unbiased approach might be necessary to identify non-mitochondrial proteins, for example additional players within the endocytosis pathway that might also regulate MDV biogenesis and mitochondrial quality control. To date, MDVs have mostly been observed in fixed cells using confocal microscopy; but live cell imaging and tracking of MDVs in tissues or 3D models will be important to understand the role of MDVs in different physiological contexts and pathologies. However, a major limitation in tracking MDVs is their small size, and advanced super-resolution optical microscopy will be relevant to image and track these ~70- to 150-nm vesicles.
Apart from cargo delivery into late endosomes/lysosomes, MDVs can also be directed to peroxisomes. Peroxisomes are the most closely linked organelle to the mitochondria and share a number of common biochemical processes. However, the purpose of MDV delivery to peroxisomes is largely unclear. A subpopulation of MDVs can also be routed to the plasma membrane. Thus, upon autophagy inhibition, MDV transport to late endosomes/lysosomes, peroxisomes and even extracellular release provides a means for recycling and secretion of mitochondrial content in order to maintain homeostasis. Moving forward, it will be critical to understand the physiological contribution and potential interdependence of canonical mitophagy, mitochondrial dynamics, and different forms of MDVs in disease pathologies where mitochondria are critical organelles including neurodegeneration, cancer, heart disease, and metabolic disorders.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
Reference
- Towers CG, Wodetzki DK, Thorburn J, et al. Mitochondrial-derived vesicles compensate for loss of LC3-mediated mitophagy. Dev Cell. 2021;56(14):2029–2042.e5.