
ABSTRACT
Minimal residual disease (MRD) refers to a low number of cells that persist anti-cancer treatment and is the major cause of relapse in solid cancers and leukemias. In chronic myeloid leukemia (CML), a paradigm for stem cell-driven cancer, MRD is maintained by tyrosine kinase inhibitor (TKI)-insensitive leukemic stem cells (LSCs), which may rely on fundamental metabolic processes to resist drug treatment. Macroautophagy/autophagy is a cytoprotective process that has been highlighted as critical for sustaining LSC survival during TKI treatment in robust experimental models of CML. Our recent study shows that the autophagy-initiating kinase ULK1 is required for maintaining energy and redox balance in CML LSCs. Pharmacological inhibition of ULK1 results in stress-induced differentiation of LSCs, rendering them sensitive to TKI treatment, uncovering a promising strategy for selective eradication of LSCs in CML patients.
Abbreviations CML: chronic myeloid leukemia; LSC: leukemic stem cell; MAPK: mitogen-activated protein kinase; MRD: minimal residual disease; TKI: tyrosine kinase inhibitor
Autophagy is a conserved catabolic mechanism that prevents the built up of damaged cellular components and facilitates the recycling of cellular material to fuel cellular energy transfer and anabolic cell growth. Normal cells, including stem cells, have evolved to utilize the autophagy process to support homeostasis and cellular health by re-using their own material. However, cancer cells can hijack autophagy for their own benefit to support uncontrolled growth or survival in harsh conditions. For example, models of pancreatic ductal adenocarcinoma, a highly aggressive lethal neoplasm of the pancreas, have revealed elevated levels of basal autophagy, required to support their rapid proliferation in a nutrient-poor environment. More commonly, several solid and liquid tumor types have been shown to upregulate autophagy flux during anti-cancer treatment to support cell survival. Among those, pancreatic ductal adenocarcinoma and colorectal cancer cells rely on autophagy as a mechanism to resist therapy during treatment with MAPK (mitogen-activated protein kinase) pathway inhibitors. The role of autophagy has also been studied in CML, a stem cell-driven blood cancer that originates in the bone marrow as a consequence of a chromosomal translocation in a single hematopoietic stem cell. This genetic aberration gives rise to the BCR-ABL oncoprotein with constitutively active tyrosine kinase activity, known to activate downstream MAPK pathways and MTOR (mechanistic target of rapamycin kinase). In CML, TKI treatment has extended life expectancy of patients, however it is generally accepted that MRD is maintained by persistent LSC which are insensitive to TKI-mediated BCR-ABL inhibition. Notably, in this clinically important cell population, cytoprotective autophagy in induced following TKI treatment.
Autophagy has become an emerging target in an effort to sensitize malignant cells to standard-of-care treatment. The anti-malarial agent chloroquine, and its derivative hydroxychloroquine (HCQ), have so far been the only available drugs that potentially could indirectly inhibit the autophagy process in cancer patients. However, HCQ targets lysosomes, organelles that fuse with autophagosomes to catalyze the last step of the autophagic process; the digestion of superfluous or damaged cellular components. In the past decade, HCQ has been used in several clinical trials including CHOICES, a phase 2 study that combined HCQ with imatinib, a first generation TKI, in CML patients with MRD. However, the results from CHOICES and other HCQ trials have indicated that maximum achievable doses of HCQ are insufficient to consistently inhibit autophagy in patients’ cancer cells. Furthermore, blocking lysosomes might not be the optimum approach and could result in undesirable side effects when used at high doses, due to their implications in other important cellular processes. This has been recognized by academia and industry, and specific pre-clinical autophagy inhibitors have been developed, including compounds active against “targetable” ATG7, PIK3C3/VPS34 and the serine-threonine kinase ULK1. Encouragingly, inhibiting the catalytic activity of ULK1, a key member of the autophagy-initiating complex, seems to be the most promising approach and results in specific and potent autophagy inhibition in standard in vitro assays.
In our study [Citation1] we demonstrate that TKI-mediated BCR-ABL inhibition leads to inhibition of MTOR activity and an increase in 5’ adenosine monophosphate-activated protein kinase (AMPK) activity in stem cell-enriched (CD34+) cells, isolated from individuals with CML. Subsequently, this leads to activation of ULK1 kinase activity, confirmed by increased phosphorylation of the ULK1 downstream target ATG13, and an increase in ULK1-dependent autophagy flux. This outcome provided the rational for investigating the impact of inhibiting ULK1 on LSC biology, alone and in combination with TKI treatment, which required development and testing of selective ULK1 inhibitors, suitable for both in vitro and in vivo studies.
We further show that inhibition of ULK1 in patient-derived CML cells affects their “stemness” by modulating mitochondrial function and central carbon metabolism pathways. Pharmacological ULK1 inhibition prevents autophagy-dependent degradation of mitochondria (mitophagy), increases mitochondrial respiration and impairs LSC’s ability to prevent accumulation of mitochondria-derived reactive oxygen species (). This correlates with enhanced cell proliferation and reduced expression of the primitive cell surface markers CD34 and PROM1/CD133, suggesting that the increase in reactive oxygen species following inhibition of ULK1 activity “pushes” LSC out of quiescence. Additionally, ULK1 inhibition has profound effects on myeloid differentiation, with human CML LSC differentiating toward more mature immunophenotypes, correlating with a significant increase in the maturation of red blood cells. This may also have clinical implication as CML is characterized by the expansion and accumulation of cells belonging to a branch of the myeloid lineage (mainly neutrophils and macrophages), causing a decrease in red blood cells and cells of the lymphoid lineage. Therefore, inhibiting ULK1 not only results in loss of TKI-insensitive LSC but also promotes the maturation of red blood cells, which could be beneficial for CML patients that present with anemia. However, what remains to be clarified is how cells with limited mitophagy can remove mitochondria, which is required for the complete erythroid maturation process. We anticipate that an alternative mechanism, such as mitochondria-derived vesicle formation, can be upregulated and responsible for mitochondria removal when mitophagy is impaired in CML cells.
Figure 1. Treatment with TKIs leads to activation of ULK1 kinase and induction of ULK1-dependent autophagy in CML LSCs. This effect contributes to therapy resistance and the persistence of bone marrow-located TKI-insensitive CML LSCs (left). MRT403 inhibits the catalytic activity of ULK1 and blocks TKI-induced autophagy in CML LSCs. The consequence of ULK1 inhibition is reduction in mitophagy, and oxidative stress-induced differentiation of primitive CML cells. This sensitizes CML LSCs to TKI treatment, supporting the concept that specific autophagy inhibition may reduce MRD when used in combination with TKI treatment.
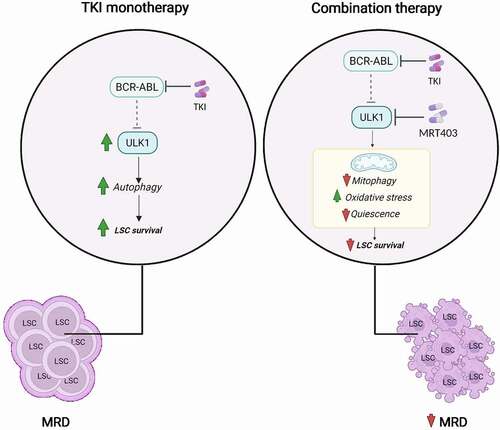
Of further clinical relevance, using both patient-derived LSC and a mouse model that resembles the human disease, we reveal a reduction in LSC when imatinib is combined with pharmacological ULK1 inhibition. Long-term transplant of murine bone marrow cells into secondary recipient mice shows that the combination treatment has a profound effect on self-renewal ability of CML LSCs, leading to gradual loss of the stem compartment. The perception that reduced stemness sensitizes leukemic cells to TKI treatment makes specific inhibition of autophagy through ULK1 inhibition an attractive approach to tackle MRD and achieve treatment-free remission in patients with CML. We are therefore committed to work with industry to develop the next generation of ULK1 inhibitors, suitable for treatment of individuals with cancer types that use autophagy as an escape route. Encouragingly, a first-in-human clinical trial combining a separate ULK1 inhibitor (DCC-3116) and MAPK pathway inhibitor has recently started for patients with rat sarcoma virus (RAS)-driven solid tumors in an advanced or metastatic phase (NCT04892017), brining further optimism that successful inhibition of autophagy in cancer patients is on the horizon.
Acknowledgments
We thank Martha M. Zarou for assistance with generating .
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
Reference
- Ianniciello A, Zarou MM, Rattigan KM, et al. ULK1 inhibition promotes oxidative stress-induced differentiation and sensitizes leukemic stem cells to targeted therapy. Sci Transl Med. 2021 Sep 29;13(613):eabd5016.