
ABSTRACT
The selective clearance of mitochondria by mitophagy is an important quality control mechanism for maintaining mitochondrial and cellular health. Iron chelation, for example by the compound deferiprone (DFP), leads to a specific form of PINK1-PRKN/Parkin-independent mitophagy; however, the molecular mechanisms underlying this are poorly understood. In our recent paper, we examined the role of the deSUMOylating enzyme SENP3 in DFP-induced mitophagy. We observed that SENP3 levels are enhanced by DFP treatment, and that SENP3 is essential for DFP-induced mitophagy. Furthermore, we identified the mitochondrial protein FIS1, which is also required for DFP-induced mitophagy, as a novel SUMO substrate. Our data demonstrate that SENP3-dependent deSUMOylation of FIS1 enhances FIS1 mitochondrial targeting, to promote mitophagy in response to DFP treatment. These findings offer new insight into the mechanisms underlying mitophagy upon iron chelation, and have relevance to the therapeutic potential of DFP in a number of disorders, including Parkinson disease. Abbreviations DFP: deferiprone; OMM: outer mitochondrial membrane. PD: Parkinson disease; SUMO: small ubiquitin like modifier.
KEYWORDS:
Main text
The selective clearance of mitochondria by mitophagy represents an important quality control mechanism in maintaining mitochondrial health. Mitochondrial damage accumulates with aging and in age-related disorders, and the importance of mitophagy is highlighted, in part, by the observation that loss of function of “classical” mitophagy mediators, such as PRKN and PINK1, leads to Parkinson disease (PD).
Iron depletion, for example with the iron chelator deferiprone (DFP), results in a specific form of mitophagy that occurs in the absence of general autophagy, and that is independent of PRKN and PINK1. However, the cellular mechanisms underlying this form of mitophagy are poorly understood, as is the contribution of this pathway to disease processes involving mitophagic clearance.
Our previous work has investigated how the post-translational modification SUMOylation orchestrates the cellular response to oxygen-glucose deprivation. SUMOylation involves the attachment of a member of the SUMO (small ubiquitin like modifier) family of proteins to lysine residues in target proteins. SUMOylation is reversible, and can be removed from target proteins by the activity of, among others, the SENP family of proteases. In particular, our work focused on the SUMO2-SUMO3-specific deSUMOylating enzyme SENP3, which has been identified as a “stress sensor” protein, whose levels can be increased or decreased by cellular stress, depending on the particular experimental paradigm used, with corresponding changes to the SUMOylation status of its substrate proteins.
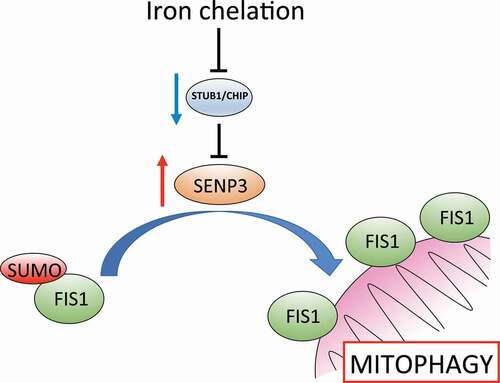
In our recent study [Citation1], we examined the mechanisms underlying DFP-induced mitophagy, and the role of SENP3-mediated deSUMOylation in this process. Upon DFP treatment of HeLa cells, we observe an increase in levels of SENP3, which occurs as a result of DFP-induced loss of the ubiquitin ligase STUB1/CHIP, which mediates proteasomal degradation of SENP3. By using a novel autophagic reporter, Mito-pHfluorin, in addition to the well-characterized Mito-Keima, we observed that cells in which SENP3 has been depleted by siRNA fail to induce mitophagy in response to DFP, identifying SENP3 as an essential mediator of this process.
We next asked as to the identity of the SENP3 substrate, or substrates, responsible for its role in DFP-induced mitophagy, and focused on FIS1, an outer mitochondrial membrane (OMM) protein that plays a critical role in stressor-induced mitophagy and had been identified as a potential SUMO substrate in a previously published proteomic screen. Like SENP3, cells lacking FIS1, either through RNAi or CRISPR-mediated knockout, fail to induce mitophagy in response to DFP, as determined by both measurements of LC3 lipidation and using the fluorescence-based reporters.
We then confirmed FIS1 as a genuine SUMO2-SUMO3 substrate, and determined that this modification takes place at a lysine residue at the extreme C terminus of FIS1 (K149). Furthermore, consistent with a role for both FIS1 and SENP3 in DFP-induced mitophagy, removal of SUMO2-SUMO3 from FIS1 is mediated by SENP3. By examining the localization of a FIS1 mutant that cannot be SUMOylated (FIS1K149R), we showed that SUMOylation of FIS1 counteracts its mitochondrial localization, with cells expressing the “constitutively deSUMOylated” FIS1K149R displaying increased mitochondrial FIS1, suggesting the SUMOylation state of FIS1 represents an important determinant of its mitochondrial targeting.
Importantly, we then examined the role of FIS1 SUMOylation in DFP-induced mitophagy. In cells expressing FIS1 fused to SUMO2, mimicking “constitutively SUMOylated” FIS1, LC3 lipidation in response to DFP is suppressed, suggesting an important role for FIS1 deSUMOylation in DFP-induced LC3-II induction. In contrast, expression of “constitutively deSUMOylated” FIS1 leads to LC3-II induction in the absence of DFP, providing a direct link between levels of LC3 lipidation, and FIS1 SUMOylation and consequent mitochondrial localization.
Finally, we asked whether the essential role for SENP3 in DFP-induced mitophagy is due to its ability to deSUMOylate FIS1. By examining both LC3-II induction and autophagosome formation with Mito-pHfluorin, we observed that while SENP3 knockdown blocks DFP-induced mitophagy, expression of “constitutively deSUMOylated” FIS1K149R restores DFP-induced mitophagy in SENP3-lacking cells. Together, these results demonstrate that SENP3 promotes DFP-induced mitophagy through promoting deSUMOylation, and thus enhances mitochondrial localization, of FIS1.
Our findings have characterized a novel modification of FIS1, SUMOylation, and suggest that this modification plays an important role in determining its mitochondrial localization. However, the mechanisms underlying this are not known. FIS1 is a tail-anchored OMM protein, and how proteins of this class are targeted to, and inserted into the membrane of mitochondria is poorly understood. FIS1 itself has also been reported to be present at the endoplasmic reticulum, peroxisomes and cytosol, and exactly how SUMOylation of its C terminus dictates the balance between these locations will likely be the subject of future work.
A growing body of evidence suggests an essential role for FIS1 in mitophagy in response to, for example, antimycin A, paraquat and DFP. FIS1 has been extensively investigated for its role in mitochondrial fission, as one of four receptors involved in the recruitment of the fission GTPase DNM1L/Drp1, itself a SUMO substrate, to the mitochondrial surface. However, DNM1L appears dispensable for mitophagy, and we observed that preventing DNM1L SUMOylation does not affect DFP-induced LC3 lipidation. Thus, the role of FIS1 in DFP-induced mitophagy likely lies outside its role in DNM1L recruitment, but the exact mechanisms underlying this remain unclear.
Interestingly, DFP is already used clinically in treatment of iron-overload disorders such as thalassemia major and has undergone phase II clinical trials for the treatment of PD, with trends toward improved motor function reported. Because defective mitophagy may be a general mechanism underlying the loss of dopaminergic neurons characteristic of PD, enhancement of mitochondrial clearance by DFP may be therapeutically beneficial. Our work highlights the potential role of SENP3 and FIS1 in these processes, and raise the possibility that pharmacologically targeting these proteins may represent a novel therapeutic strategy in PD.
In conclusion, we have investigated mitophagy induced by the iron chelator DFP, and identified the essential roles of the deSUMOylating enzyme SENP3, and the OMM protein FIS1 in this process, through SUMO-mediated regulation of FIS1 mitochondrial localization (). Our findings both enhance understanding of this novel mitophagy pathway, provide molecular insight into the therapeutic potential of DFP, and highlight these proteins as possible targets in disorders in which defective mitophagy is implicated.
Figure 1. Iron chelation with deferiprone leads to reduced protein levels of the ubiquitin ligase STUB1/CHIP, which promotes degradation of the deSUMOylating enzyme SENP3. Reduced STUB1/CHIP levels lead to stabilization of SENP3, favoring deSUMOylation of its substrate FIS1. FIS1 deSUMOylation leads to an enhancement of FIS1 localization at the outer mitochondrial membrane, permitting the induction of mitophagy.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Correction Statement
This article has been corrected with minor changes. These changes do not impact the academic content of the article.
Additional information
Funding
Reference
- Waters E, Wilkinson KA, Harding AL, et al. The SUMO protease SENP3 regulates mitochondrial autophagy mediated by Fis1. EMBO Rep. 2022;23(2):e48754.