
ABSTRACT
The discovery of recurrent mutations in subunits and regulators of the vacuolar-type H+-translocating ATPase (V-ATPase) in follicular lymphoma (FL) highlights a role for macroautophagy/autophagy, amino-acid, and nutrient-sensing pathways in the pathogenesis of this disease. Here, we report on novel mutations in the ER-resident chaperone VMA21, which is involved in V-ATPase assembly in 12% of FL. Mutations in a novel VMA21 hotspot (p.93X) result in the removal of a C-terminal non-canonical ER retrieval signal thus causing VMA21 mislocalization to lysosomes. The resulting impairment in V-ATPase activity prevents full lysosomal acidification and function, including impaired pH-dependent protein degradation as shown via lysosomal metabolomics and ultimately causes a degree of amino acid depletion in the cytoplasm. These deficiencies result in compensatory autophagy activation, as measured using multiple complementary assays in human and yeast cells. Of translational significance, the compensatory activation of autophagy creates a dependency for survival for VMA21-mutated primary human FL as shown using inhibitors to ULK1, the proximal autophagy-regulating kinase. Using high-throughput microscopy-based screening assays for autophagy-inhibiting compounds, we identify multiple clinical grade cyclin-dependent kinase inhibitors as promising drugs and thus provide new rationale for innovative clinical trials in FL harboring aberrant V-ATPase.
Abbreviations: ALs: autolysosomes; APs: autophagosomes; ER: endoplasmic reticulum; FL: follicular lymphoma; GFP: green fluorescent protein; IP: immunoprecipitation; LE/LY: late endosomes/lysosomes; Lyso-IP: lysosomal immunoprecipitation; OST: oligosaccharide transferase; prApe1: precursor aminopeptidase I; SEP: super ecliptic pHluorin; V-ATPase: vacuolar-type H+-translocating ATPase
Introduction
Follicular lymphoma (FL) constitutes the most common indolent B-cell lymphoma, with an incidence and prevalence of ~15,000 and ~100,000 cases, respectively, in the US [Citation1]. FL remains incurable with conventional therapies and most patients receive multiple treatment regimens over the course of their illness. While the overall clinical outlook for FL patients is improving, those with short remission durations following upfront chemoimmunotherapy, or patients transforming to large B cell lymphoma have substantially shortened overall survival [Citation2]. A better understanding of FL growth and survival dependencies and the development of novel targeted FL therapies therefore remain a research priority [Citation3–5].
The identification of MTOR-activating mutations in the small G-protein RRAGC, a component of the amino-acid signaling pathway to MTOR, has highlighted an opportunity to target this pathway in FL [Citation6–8]. The RRAGC protein is part of a macromolecular complex consisting at a minimum of the V-ATPase, the amino acid sensor/transporter SLC38A9, a pentameric protein complex termed Ragulator (comprised of LAMTOR1 to LAMTOR5), FLCN (folliculin) and FNIP2, the small G-proteins RRAGA to RRAGD (as heterodimers) and MTORC1 that exists on the lysosomal surface [Citation9–16]. This complex dynamically regulates autophagy, nutrient sensing, and the activity of MTORC1 and AMP-activated protein kinase/AMPK.
Recently, multiple groups have reported on heterozygous hotspot mutations in the V-ATPase V1 subunit ATP6V1B2, an evolutionarily conserved enzyme, in 10% – 15% of FL [Citation6,Citation17,Citation18]. The V-ATPase is a proton pump and is primarily responsible for lysosomal acidification [Citation19]. The ATP6V1B2 subunit is one of two principal components of the ring-like hexameric cytoplasmic V1 component of the V-ATPase, which is responsible for ATP hydrolysis, while the V0 component is membrane bound, constitutes the stalk and forms the proton channel of the V-ATPase holoenzyme [Citation19–21]. Full acidification of lysosomes is needed for activation of protein and peptide hydrolases that degrade protein cargo delivered to lysosomes via autophagosomes, multivesicular bodies, microautophagy or macropinocytosis [Citation22]. The mutations in ATP6V1B2 result in a hypomorphic V-ATPase with reduced ability for lysosomal acidification, impaired protein degradation and a resulting strong compensatory upregulation of autophagic flux. The upregulation of autophagy is important for FL B cell survival as the treatment of ATP6V1B2 mutant human FL B cells with inhibitors of ULK1 (unc-51 like autophagy activating kinase 1), the apical autophagy initiating kinase, results in FL cell death.
Various other genes related to V-ATPase function, including ATP6AP1 and ATP6AP2 are mutated in FL but the functional consequences of these mutations on FL biology and autophagy or MTOR regulation have not been elucidated [Citation6]. The V-ATPase V0 multiprotein transmembrane complex is assembled in the ER with the obligate involvement of a set of assembly chaperones [Citation23–26]. These chaperones include ATP6AP1 and ATP6AP2 as well as the small protein VMA21. Here, we report on common mutations in VMA21 in FL that result in aberrant VMA21 cellular trafficking, and reduced V-ATPase holoenzyme function with an associated inability to fully acidify lysosomes. This lysosomal defect is associated with an aberrant lysosomal and cytosol metabolome and a strong compensatory activation of autophagy. These novel findings therefore unify the phenotypes of VMA21 and ATP6V1B2 mutant FL thus extending the pathobiological consequences of hypomorphic V-ATPase pathology to a combined total of 20–25% of FL.
We previously demonstrated a survival dependence of ATP6V1B2 mutant FL on autophagic flux, which motivated us to screen for drugs that inhibit VMA21-mutant induced autophagy. Our high-throughput screen of 4,577 compounds identified multiple compound classes with autophagy inhibitory properties, most prominently cyclin-dependent kinase inhibitors. The identification of compounds that are in clinical development or approved for clinical use in humans provides a direct path to novel therapeutic approaches in FL. Altogether, we report that a substantial subset of FL carry mutations that through various mechanisms result in hypofunctional V-ATPase, an elevated lysosomal pH and strong compensatory activation of autophagy, which lays the mechanistic foundation for innovative clinical trials repurposing various promising autophagy inhibitory drugs as therapeutics for V-ATPase-mutant FL.
Results
The frequency and nature of VMA21 mutations in follicular lymphoma
Based on recurrent common mutations in genes associated with V-ATPase biology (ATP6V1B2, ATP6AP1, ATP6AP2), we re-sequenced the three coding exons of the V-ATPase chaperone gene VMA21 and adjacent intronic regions using Sanger sequencing in highly pure FL B cell DNA isolated from flow sorted FL B cells in a total of 151 FL grades 1–3A and 19 transformed FL cases [Citation6,Citation17,Citation18]. We confirmed the somatic nature of all sequence variants using paired sorted lymph-node or blood- derived CD3+ T cell DNA. We identified 12% (18/151) of FL cases and 2/19 t-FL with non-synonymous VMA21 mutations, most of which were hemizygous ().
Figure 1. Schema of the VMA21 protein and details of novel follicular lymphoma (FL)-associated VMA21 mutations. (A) Schema of the VMA21 protein comprising 101 amino acids and novel mutations detected in FL. The VMA21 protein is recurrently targeted by a nonsense mutation (p.93X) resulting in a shortened protein of 92 amino acids. Blue: transmembrane domains. (B) Sanger sequence tracings. T = lymphoma B cells, N = paired CD3+ T cells. (C) Schema of the chaperone function of WT VMA21 toward V-ATPase assembly in the ER/Golgi.
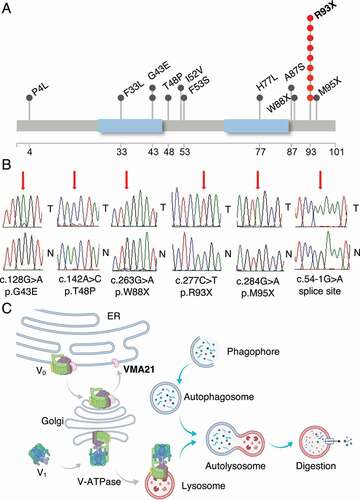
The VMA21 gene encodes a small transmembrane ER/Golgi chaperone protein of 101 amino acids. Topologically, the N and C termini are cytoplasmic and two transmembrane segments connect via a lumenal (ER, Golgi, lysosome) domain. The VMA21 gene is targeted in FL by a hotspot nonsense mutation (c.C277T; p.R93X) comprising 40% of all detected mutations resulting in the deletion of the C-terminal nine amino acids (amino acids 93–101). Additional nonsense mutations (p.W88X and p.W95X) also delete parts of the C terminus. Various missense mutations targeted other parts of the gene ().
A review of gene mutations co-occurring in FL cases with mutant VMA21 demonstrated that mutations in other genes related to V-ATPase function and autophagy, including ATP6V1B2, ATP6AP1, ATP6AP2, and sporadic mutations in ATP6V0A4, ATP6V0C and ATP6V1A were absent in VMA21 mutant cases. Furthermore, the majority of VMA21 mutant cases carried mutations in KMT2D and CREBBP (Figure S1).
Of the VMA21 mutations reported here, eleven were detected in males and one in a female while the sex of the patients with mutations detected in samples from the de-identified FL cohort II is unknown. Further, five VMA21 mutations were detected in untreated FL patients and seven in relapsed patients, while the clinical status of the patients with mutations detected in the de-identified samples is unknown.
A review of CRISPR-targeting data based on 1027 cell lines reported in the depmap portal (https://depmap.org/portal/) indicated that VMA21 disruption conferred a competitive disadvantage on cells (categorized as imparting reduced cell fitness). The effect, however, was less pronounced when compared with disruption of ATP6AP1 or ATP6AP2, which were essential for most of the cell lines tested.
FL-associated VMA21 mutations activate autophagic flux
Elevated LC3-II levels in VMA21 mutant (MUT) cells
Given the strong autophagy-activating phenotype of mutations in the V-ATPase subunit ATP6V1B2, we tested for effects of VMA21 mutations on autophagy using the steady state levels of the autophagosomal marker LC3-II [Citation27]. We generated stable HEK293T cells carrying the doxycycline-inducible vector pCW57.1 alone or vector containing cDNAs for WT or MUT (p.93X) HA-tagged VMA21. Upon transgene induction, we detected strongly elevated levels of LC3-II in the mutant transfectants (, B). We also performed experiments with the V-ATPase and autophagy inhibitor bafilomycin A1 and found that LC3-II levels in the mutant increased further, in agreement with elevated autophagy flux (, B).
Figure 2. Follicular lymphoma-associated VMA21 mutations activate autophagic flux. (A) HEK293T cells transfected with empty vector, WT, or mutant (93X) VMA21 treated -/+ bafilomycin A1. Immunoblots of the indicated antigens. (B) Densitometry quantification (LC3-II:ACTB) of n = 4 independent experiments from representative panel A. Statistical comparisons: i) VMA21 93X versus WT (A; lanes 5–6). ii) VMA21 93X versus WT plus -/+ bafilomycin A1. (A; lanes 8–9), using unpaired 2-tailed t-testing and Bonferroni corrections (*: p < 0.05; **: p < 0.01). Bars: standard deviations. (C) Electron microscopy and enumeration of autophagosomes (AP), autolysosomes (AL) and late endosomes/lysosomes (LE/LY) in HEK293T cells expressing VMA21 WT or MUT 93X. Representative images of N = 70–90 each. (D) Statistical comparisons of data generated in panel C using unpaired 2-tailed Mann-Whitney U-test and Bonferroni corrections. ***: p < 0.001). Bars: Standard eviations. (E-K) S. cerevisiae cells expressing either Vma21 (WT) or Vma21[∆66-77] were analyzed for autophagy. The cells were grown in YPD (-N, 0 min) to mid-log phase, and shifted to nitrogen starvation (SD-N) (E) The GFP-Atg8 processing assay. Pgk1 was used as a loading control. (F) The ratio of free GFP to Pgk1 after 45 min SD-N treatment was quantified. Mean ± SD of n = 4 independent experiments. Unpaired, 2-tailed t-test; *: p < 0.05. (G) Pho8∆60 enzymatic activity was measured under growing and nitrogen-starvation conditions. Mean ± SD of n = 5 independent experiments. Two-way ANOVA; ***: p < 0.001. (H) The prApe1 processing assay. Dpm1 was used as a loading control. (I) The ratio of Ape1 to prApe1 after 15 min SD-N treatment was quantified. Mean ± SD of n = 3 independent experiments. Unpaired, 2-tailed t-test; *: p < 0.05. (J) The Atg8 lipidation assay. Pgk1 was used as a loading control. (K) The ratio of Atg8–PE to Pgk1 after 45 min of nitrogen starvation was quantified. Mean ± SD of n = 4 independent experiments. Two-way ANOVA; *: p < 0.05, **: p < 0.01.
![Figure 2. Follicular lymphoma-associated VMA21 mutations activate autophagic flux. (A) HEK293T cells transfected with empty vector, WT, or mutant (93X) VMA21 treated -/+ bafilomycin A1. Immunoblots of the indicated antigens. (B) Densitometry quantification (LC3-II:ACTB) of n = 4 independent experiments from representative panel A. Statistical comparisons: i) VMA21 93X versus WT (A; lanes 5–6). ii) VMA21 93X versus WT plus -/+ bafilomycin A1. (A; lanes 8–9), using unpaired 2-tailed t-testing and Bonferroni corrections (*: p < 0.05; **: p < 0.01). Bars: standard deviations. (C) Electron microscopy and enumeration of autophagosomes (AP), autolysosomes (AL) and late endosomes/lysosomes (LE/LY) in HEK293T cells expressing VMA21 WT or MUT 93X. Representative images of N = 70–90 each. (D) Statistical comparisons of data generated in panel C using unpaired 2-tailed Mann-Whitney U-test and Bonferroni corrections. ***: p < 0.001). Bars: Standard eviations. (E-K) S. cerevisiae cells expressing either Vma21 (WT) or Vma21[∆66-77] were analyzed for autophagy. The cells were grown in YPD (-N, 0 min) to mid-log phase, and shifted to nitrogen starvation (SD-N) (E) The GFP-Atg8 processing assay. Pgk1 was used as a loading control. (F) The ratio of free GFP to Pgk1 after 45 min SD-N treatment was quantified. Mean ± SD of n = 4 independent experiments. Unpaired, 2-tailed t-test; *: p < 0.05. (G) Pho8∆60 enzymatic activity was measured under growing and nitrogen-starvation conditions. Mean ± SD of n = 5 independent experiments. Two-way ANOVA; ***: p < 0.001. (H) The prApe1 processing assay. Dpm1 was used as a loading control. (I) The ratio of Ape1 to prApe1 after 15 min SD-N treatment was quantified. Mean ± SD of n = 3 independent experiments. Unpaired, 2-tailed t-test; *: p < 0.05. (J) The Atg8 lipidation assay. Pgk1 was used as a loading control. (K) The ratio of Atg8–PE to Pgk1 after 45 min of nitrogen starvation was quantified. Mean ± SD of n = 4 independent experiments. Two-way ANOVA; *: p < 0.05, **: p < 0.01.](/cms/asset/dfc76a98-26e9-4d1c-ba05-8e46b4cb8486/kaup_a_2050663_f0002_b.gif)
Elevated numbers of autolysosomes in VMA21 MUT cells as measured by electron microscopy
One of the “gold standard” assays to quantify autophagic flux, here defined as the generation of autophagosomes (APs) and their subsequent maturation and fusion with lysosomes, is via the enumeration of APs, autolysosomes (ALs) and late endosomes/lysosomes (LE/LY) in cells based on electron microscopy (EM). Using the inducible HEK293T VMA21p.93X MUT or VMA21 WT cell lines we counted APs, ALs and LE/LYs in 70–90 EM images each per cell line. We found that the number of ALs was substantially and significantly elevated in the VMA21p.93X cell lines with a mean of 0.23 for vector, 0.25 for WT and 1.06 for MUT per cell, confirming pathological elevation of autophagic flux induced by the MUT VMA21 protein (, D).
Elevated autophagic flux in yeast carrying the Vma21[∆66-77] mutation in the genome
To further investigate the effect of the VMA21p.93X mutation on autophagy and autophagic flux, we proceeded to test its effects in a yeast model. Sequence alignment showed that the VMA21p.93X mutation corresponds to yeast Vma21[∆66-77]. Therefore, we generated a Vma21[∆66-77] mutation in the genome. As a readout of autophagy activity, we measured green fluorescent protein (GFP)-Atg8 processing in S. cerevisiae cells expressing either full-length Vma21 (WT) or the truncated version Vma21[∆66-77]. Atg8 binds to both sides of the phagophore [Citation28], and a portion of Atg8, or GFP-tagged Atg8, is delivered to the vacuole via autophagy; GFP is relatively resistant to vacuolar hydrolases and the free form can be resolved and detected by western blot. The Vma21[∆66-77] mutant showed an increased level of free GFP under nitrogen-starvation conditions, suggesting elevated autophagic flux in the mutant cells (, F).
We extended our measurement of autophagy to the quantitative Pho8∆60 assay. Upon deletion of the first 60 amino acids at the N terminus, Pho8 phosphatase can only be delivered to the vacuole nonselectively through autophagy [Citation29]. Subsequently, the Pho8∆60 propeptide gets cleaved by vacuolar hydrolases, thereby activating its phosphatase activity. We observed a significant increase in autophagy-dependent Pho8∆60 phosphatase activity under nitrogen-starvation conditions in the Vma21[∆66-77] mutant cells ().
Next, we performed one additional assay that monitors a type of selective autophagy. Ape1 is a vacuolar hydrolase that is originally synthesized as a cytosolic precursor form (prApe1). The prApe1 can be delivered to the vacuole through either macroautophagy or the cytoplasm-to-vacuole targeting/Cvt pathway [Citation30]. In the vacuole, prApe1 is processed to a mature form with lower molecular mass that can be easily detected by western blot. The assay was performed in cells with a vac8∆ background, which prevents vacuolar delivery of prApe1 under growing conditions; however, the precursor is efficiently delivered via autophagy under nitrogen-starvation conditions. Unlike the previous two assays, import of prApe1 depends on selectivity factors including the receptor Atg19 and the scaffold Atg11. We observed a similar autophagy increase in the Vma21[∆66-77] mutant cells under starvation conditions (, I).
To analyze an earlier aspect of autophagy, we examined the induction step, following the endogenous Atg8 protein through a lipidation assay. The lipidated form, Atg8–PE, displays a faster electrophoretic mobility relative to nonlipidated Atg8 that can be detected by western blot. The cells expressing Vma21[∆66-77] showed a substantial increase in the levels of Atg8–PE under both growing and nitrogen-starvation conditions, indicating upregulated autophagy induction (, K).
Generation of quantitative autophagic flux reporter cell lines expressing inducible VMA21 WT or MUT
We generated HEK293T cell lines stably expressing the well-studied cleavable autophagy reporter fusion protein GFP-LC3-(ATG4 proteolytic cleavage site)-RFP together with inducible HA-VMA21 WT or MUT () [Citation31]. Upon autophagy induction, the GFP-LC3-RFP fusion protein is cleaved into GFP-LC3 and RFP, followed by lipid conjugation of LC3 and association of GFP-LC3-II with APs and ALs. We employed these cells for a variety of autophagy assays and for chemical autophagy inhibitor screens detailed below.
Figure 3. Adaptation of an autophagic reporter assays to measure VMA21 MUT activated autophagy. (A) Schema of the autophagy reporter fusion protein GFP-LC3-(ATG4 cleavage site)-RFP used to make stable HEK293T cells also carrying inducible VMA21 WT and MUT. The less acidic lysosomes in VMA21 MUT cells (see Results) allow for persistence of green fluorescence autolysosomes as a quantitative readout for autophagy. (B-C) Detection of free GFP generated in lysosomes in stable HEK293T or stable OCI-LY1 lymphoma cells also carrying inducible VMA21 WT and MUT. Immunoblots for the indicated antigens. (D) Confocal microscopy images of stable HEK293T cells also carrying inducible empty vector, or VMA21 WT and MUT. (-) empty vector, SAR405: a PIK3C3/VPS34 inhibitor, MRT68921: a ULK1 inhibitor. (E) Densitometry quantification (free GFP:ACTB) of n = 3 independent experiments from representative panel B. Statistical comparisons: i) ATP6V1B2 MUT 371C or 400Q versus WT (B: lanes 8–10) or VMA21 93X versus WT (B: lanes 11–12), using unpaired 2-tailed t-testing and Bonferroni corrections (*: p < 0.05; **: p < 0.01, ***: p < 0.001). (F) Densitometry quantification (free GFP:ACTB) of three independent experiments from representative panel C. Statistical comparisons: i) ATP6V1B2 MUT 400Q versus WT (B: lanes 7–8) or VMA21 93X versus WT (C lanes 9–10). Statistical comparisons as in panel E. (G) Mean GFP-positive vesicles per cells from images as in panel D. (-) vector, VMA21 WT or MUT 93X, or VMA21 p.93X pretreated with the autophagy inhibitors SAR405 or MRT68921. Statistical comparisons using unpaired 2-tailed t-testing and Bonferroni corrections (*: p < 0.05; **: p < 0.01, ***: p < 0.001). Bars: tandard deviations in panels E-G.
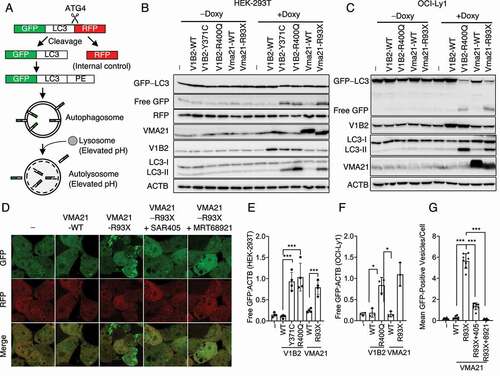
Elevated levels of free GFP in VMA21 MUT cells carrying GFP-LC3-RFP autophagy reporters
Upon association of GFP-LC3-II with phagophores and subsequent fusion of APs with lysosomes, the GFP-LC3-II protein is subjected to partial proteolysis by lysosomal proteases resulting in free GFP. We therefore measured free GFP in HEK293T cells and OCI-LY1 lymphoma cells inducibly expressing WT or MUT VMA21 or WT or MUT ATP6V1B2 (p.371C and/or p.400Q) as controls. We detected elevated levels of endogenous LC3-II and elevated levels of free (lysosomal) GFP in all MUT cell lines (, C and quantification in E, F).
Elevated numbers of autolysosomes in VMA21 MUT cells and inhibition by ULK1 or PIK3C3/VPS34 inhibitors
Following induction of VMA21 MUT expression in the reporter cells, we detected many green cytoplasmic vesicles (APs and ALs) and much fewer such vesicles in VMA21 WT cells indicative of VMA21-MUT induced autophagy (). The GFP fluorescence signal is usually quenched in acidic lysosomes over time. However, as detailed below, the lysosomes in VMA21 MUT cells are not as acidic as in WT cells thus allowing for persistence of GFP protein and the green ALs as shown in .
Next, we tested the effects of two well-studied small molecule inhibitors of the proximal autophagy initiator proteins ULK1 (MRT68921) and PIK3C3/VPS34 (SAR405) on VMA21-MUT induced autophagic flux [Citation32,Citation33]. As can be seen in –G, both compounds substantially inhibited the appearance of APs and ALs in VMA21 MUT cells, thus demonstrating involvement of canonical macroautophagy initiation complexes in VMA21-MUT activated autophagy.
The mutant VMA21 protein mislocalizes to lysosomes because of a deleted C-terminal endoplasmic reticulum (ER) retrieval signal sequence
Results from VMA21 cellular localization studies using confocal microscopy
Yeast Vma21 is one of five proteins known to be necessary for successful V-ATPase V0 complex assembly in the ER [Citation23,Citation34]. Vma21 is an assembly factor and a chaperone for the V0 complex, accompanying it as it traffics from the ER toward the Golgi, although mechanistic details on how this is accomplished are lacking. Upon arrival at the Golgi, Vma21 dissociates from the V0 complex and Vma21 is shuttled back to the ER via a C-terminal KKXX (KKED in yeast Vma21) retrieval sequence [Citation23].
To study the functional consequences of the deletion of the C terminus (amino acids 93–101) in human MUT VMA21, we employed confocal microscopy to localize HA-tagged WT and MUT VMA21 in cells. We found that WT VMA21 predominantly localized to the ER/Golgi, whereas MUT VMA21 was strongly enriched in lysosomes as evidenced by enhanced colocalization of MUT VMA21 with the lysosomal protein LAMP1 (, B).
Figure 4. HA-VMA21 MUT 93X proteins aberrantly localized to lysosomes. (A) Confocal microscopy and merged images of cells stained for CANX (ER), GOLGA2 (Golgi) and LAMP1 (lysosomes) and HA-VMA21 WT or MUT. (B) Pearson correlation coefficients for colocalization of VMA21 WT or MUT proteins from (A). (C) Representative results for lysosomal-IPs (lyso-IPs; see Materials and Methods) out of stable HEK293T cells expressing the fusion protein bait 3xHA-TMEM192 or 2xFLAG-TMEM (negative control) and inducible VMA21 WT and MUT. Detection of protein in direct cell lysates and in the retained lysosomal IP preparations for the indicated antigens. (D) Densitometry quantification (VMA21 MUT:WT protein) of n = 3 independent experiments from representative panel C and quantification of V-ATPase components from either lysates or enriched lysosomes. Statistical comparisons using unpaired 2-tailed t-testing (*: p < 0.05). Bars: Standard deviations. (E-H) S. cerevisiae cells expressing either full-length Vma21 (WT) or the truncation mutant Vma21[∆66-77] were analyzed for assembly of V-ATPase subunits. (E) The cellular and vacuolar Vma2 protein levels were monitored: The cells were grown in YPD to mid-log phase, and vacuoles were isolated. The levels of Vma2 were analyzed by western blot. Ponceau S staining was used for total protein normalization. (F) The cellular and vacuolar Vma4 protein levels were monitored as in (E). (G) Vma2 and (H) Vma4 protein levels. Either cellular or vacuolar Vma2 and Vma4 protein levels in WT cells were set to 100%, and the corresponding levels in Vma21[∆66-77] cells were normalized. Mean ± SD of n = 3 independent experiments are shown. Unpaired, 2-tailed t-test; *: p < 0.05, ns: not significant.
![Figure 4. HA-VMA21 MUT 93X proteins aberrantly localized to lysosomes. (A) Confocal microscopy and merged images of cells stained for CANX (ER), GOLGA2 (Golgi) and LAMP1 (lysosomes) and HA-VMA21 WT or MUT. (B) Pearson correlation coefficients for colocalization of VMA21 WT or MUT proteins from (A). (C) Representative results for lysosomal-IPs (lyso-IPs; see Materials and Methods) out of stable HEK293T cells expressing the fusion protein bait 3xHA-TMEM192 or 2xFLAG-TMEM (negative control) and inducible VMA21 WT and MUT. Detection of protein in direct cell lysates and in the retained lysosomal IP preparations for the indicated antigens. (D) Densitometry quantification (VMA21 MUT:WT protein) of n = 3 independent experiments from representative panel C and quantification of V-ATPase components from either lysates or enriched lysosomes. Statistical comparisons using unpaired 2-tailed t-testing (*: p < 0.05). Bars: Standard deviations. (E-H) S. cerevisiae cells expressing either full-length Vma21 (WT) or the truncation mutant Vma21[∆66-77] were analyzed for assembly of V-ATPase subunits. (E) The cellular and vacuolar Vma2 protein levels were monitored: The cells were grown in YPD to mid-log phase, and vacuoles were isolated. The levels of Vma2 were analyzed by western blot. Ponceau S staining was used for total protein normalization. (F) The cellular and vacuolar Vma4 protein levels were monitored as in (E). (G) Vma2 and (H) Vma4 protein levels. Either cellular or vacuolar Vma2 and Vma4 protein levels in WT cells were set to 100%, and the corresponding levels in Vma21[∆66-77] cells were normalized. Mean ± SD of n = 3 independent experiments are shown. Unpaired, 2-tailed t-test; *: p < 0.05, ns: not significant.](/cms/asset/01aca9c6-103d-47bb-b707-2d7c54177ec2/kaup_a_2050663_f0004_oc.jpg)
How does human WT VMA21 cycle back to the ER? The human VMA21 protein lacks a typical COPI complex (a vesicle coating protein complex comprising seven subunits) recognition signal at the C terminus (KKXX or KXKXX), which is needed to transport proteins back from the cis-Golgi to the ER, but instead carries the sequence R93QWREGKQD101. We proceeded to test if the sequence KQD was sufficient to serve as an ER retrieval signal and if its deletion could mimic the phenotype of the VMA21p.93X mutant. We generated the VMA21 MUTs p.K99A and p.delKQD(99–101) and analyzed their intracellular localization. Like VMA21 p.93X, both p.K99A and p.delKQD(99–101) MUTs enriched in lysosomes, thus providing novel evidence that the VMA21 C-terminal KQD sequence is functional as an atypical ER retrieval signal (Figure S2A, C). Both mutants also activated autophagy as detected in the GFP-LC3-RFP reporter cell lines (Figure S2B, D).
Results from VMA21 localization studies using lysosomal immunoprecipitation (lyso-IP)
Given the central roles of the lysosome in V-ATPase biology, MTOR and autophagy regulation and its emerging role in MUT VMA21 biology, we employed whole-lysosome immunoprecipitation by following the methods published by Abu-Remaileh et al. [Citation35]. We generated HEK293T cells stably expressing the triple HA-tagged lysosomal membrane protein TMEM192 (3xHA-TMEM192) as a bait together with inducible WT or MUT VMA21. Following induction of VMA21 expression, we immunoprecipitated lysosomes with anti-HA-beads added in limiting amounts.
Next, we measured the amount of selected organelle indicator proteins (CANX [calnexin], ER; GOLGA2/GM130, Golgi; ACTB, cytosol; LAMP1, lysosomes) as well as ATP6V0, ATP6V1 and VMA21 in the post-IP supernatant and in the lysosomal preparations via immunoblotting (, D). We confirmed that the lyso-IP method enriched for lysosomes and, importantly, that MUT VMA21 (which runs faster than WT on SDS-PAGE gels) was enriched in lysosomes. Of additional interest, the amount of ATP6V0 or ATP6V1 proteins in lysosomes was not altered.
To determine if the Vma21[∆66-77] mutation affects V-ATPase assembly in yeast, we measured vacuolar protein levels of V-ATPase subunits Vma2 and Vma4, which are homologous to human ATP6V1B1 and ATP6V1E1, respectively. Vacuoles were purified from logarithmically grown yeast cells by dextran lysis and flotation, followed by western blotting. Cellular levels of Vma2 and Vma4 were not significantly changed between WT and Vma21[∆66-77] cells. Vma2 was present at a slightly lower amount in Vma21[∆66-77] mutant vacuoles (, G), whereas vacuolar Vma4 levels were not significantly changed between Vma21[∆66-77] mutant and WT cells (, H).
An aberrant VMA21-MUT associated proteome defined via immunoprecipitation (IP)-mass spectrometry
We conducted two IP-mass spectrometry experiments to define the VMA21 WT- and MUT-protein associated interactome. Using OCI-LY1 lymphoma cells expressing inducible HA-tagged VMA21 WT or MUT or cells carrying vector alone, we immunoprecipitated out of detergent cell lysates using anti-HA beads and proceeded with quantitative tandem mass spectrometry and peptide identification. Following data normalization incorporating the normalized spectral absorbance factor/NSAF and the relative amounts of immunoprecipitated VMA21 WT or MUT proteins, we detected a greater amount of almost all V-ATPase protein subunits (V0 + V1) in the VMA21 MUT IPs compared with VMA21 WT (, B). The only exception was ATP6V0C, the central c subunit of the membrane-spanning V-ATPase V0 complex, of which slightly less precipitated with MUT VMA21 when compared with WT VMA21. The latter finding is likely a reflection of the VMA21 MUT protein’s inability to cycle back to the ER, resulting in reduced binding and assembly of additional V-ATPase V0 complexes and thus a net decrease in the sum of ATP6V0C bound to VMA21 complexes existing in the ER, Golgi, and post-Golgi compartments.
Figure 5. Results from IP-mass spectrometric analysis of proteins co-immunoprecipitating with HA-VMA21 WT or MUT. (A) The cryo-EM structure of human V-ATPase (ProteinDataBankID 6WM2,Mol* [Citation36] and RCSB PDB) as published by Wang et al. in 2020 [Citation26]. (B) Heatmap display of V-ATPase proteins co-immunoprecipitating with HA-VMA21 MUT or WT proteins based on n = 2 independent experiments. The data in B-D are normalized incorporating the normalized spectral absorbance factor (NSAF) and the relative amounts of immunoprecipitated VMA21 WT or MUT proteins. Displayed is the log2 of MUT:WT protein amounts. (C) Heatmap display of COPI complex proteins co-immunoprecipitating with HA-VMA21 MUT or WT proteins. (D) Heatmap display of DDOST (dolichyl-diphosphooligosaccharide—protein glycosyltransferase non-catalytic subunit) complex proteins co-immunoprecipitating with HA-VMA21 MUT or WT proteins.
![Figure 5. Results from IP-mass spectrometric analysis of proteins co-immunoprecipitating with HA-VMA21 WT or MUT. (A) The cryo-EM structure of human V-ATPase (ProteinDataBankID 6WM2,Mol* [Citation36] and RCSB PDB) as published by Wang et al. in 2020 [Citation26]. (B) Heatmap display of V-ATPase proteins co-immunoprecipitating with HA-VMA21 MUT or WT proteins based on n = 2 independent experiments. The data in B-D are normalized incorporating the normalized spectral absorbance factor (NSAF) and the relative amounts of immunoprecipitated VMA21 WT or MUT proteins. Displayed is the log2 of MUT:WT protein amounts. (C) Heatmap display of COPI complex proteins co-immunoprecipitating with HA-VMA21 MUT or WT proteins. (D) Heatmap display of DDOST (dolichyl-diphosphooligosaccharide—protein glycosyltransferase non-catalytic subunit) complex proteins co-immunoprecipitating with HA-VMA21 MUT or WT proteins.](/cms/asset/be072e9a-5885-4954-b109-0b6170db8b47/kaup_a_2050663_f0005_oc.jpg)
In addition, the V-ATPase accessory subunits ATP6AP1 and ATP6AP2, which are required for V-ATPase V0 assembly, also co-precipitated with VMA21 WT and MUT. The proteins ATP6AP1 and ATP6AP2, which are both mutated in FL, are components of the V-ATPase V0 complexes and are located centrally within the V0 membrane-spanning ring formed by nine ATP6V0C/V0-c subunits () [Citation20,Citation21,Citation26].
We also inspected the VMA21 WT- and MUT-associated proteome for other interactions that could shed light on their functions. Importantly, we detected the COPI complex (comprising the subunits COPA, COPB1, COPB2, ARCN1/COPD, COPE, COPG1, COPG2) in the IPs further supporting the involvement of this trafficking complex in VMA21 and V-ATPase biology (please note that only small or no amounts of COPE were recovered and thus are omitted from the display in ). Of note, less COPI subunit proteins (five out of six) precipitated with MUT than WT VMA21 supporting prior conclusions of impaired COPI interaction with VMA21 MUT proteins (), although effects of additional proteins remain possible.
Multiple other proteins with interesting functions co-precipitated with VMA21 and could be the subject for future studies (Tables S1-2). For instance, we detected subunits (DDOST, RPN1, RPN2, DAD1, and STT3A) of the ER resident oligosaccharide transferase complex (OST), most of which precipitated strongly and highly preferentially with the VMA21 MUT protein (). Given the essential role that OST serves in the protein complex that adds complex high mannose oligosaccharides to nascent ER-resident proteins and the observed glycosylation defects in patients carrying germline mutations in VMA21, ATP6AP1 and ATP6AP2, it is possible that the enhanced VMA21 MUT-OST interaction impairs such glycosylation possibly through aberrant OST sequestration [Citation37].
Mutant VMA21 confers a defect on the V-ATPase holoenzyme proton pump resulting in elevated lysosomal pH
We previously showed that MUT ATP6V1B2 results in a hypomorphic V-ATPase enzyme and higher lysosomal pH. We therefore proceeded with measurements of lysosomal pH in stably transduced HEK293T cells expressing WT or MUT VMA21. Cells were loaded with a pH-sensitive dual reporter dye conjugated to dextran and the cells were subsequently analyzed using a fluorescence-activated cell sorter. As controls, cells in parallel were briefly incubated with buffers at a defined pH of 3.5, 4.5 or 5.5, or were treated with bafilomycin A1 to completely inhibit V-ATPase function (). Whereas treatment with bafilomycin A1 resulted in an elevated lysosomal pH as expected from its ability to inhibit the V-ATPase, we detected an acidic lysosomal pH in the range of 4.2 in VMA21 WT cells. We measured a less acidic lysosomal pH in the range of >4.5–5.0 in the VMA21 MUT cell lines and in cells expressing a VMA21-targeted shRNA but not a scrambled shRNA. Therefore, the VMA21 MUT caused a partial lysosomal acidification defect resulting in a modestly elevated lysosomal pH (, B).
Figure 6. Follicular-lymphoma associated VMA21 mutations reduce the ability of the V-ATPase to acidify lysosomes because of reduced lysosomal V-ATPase activity. (A) Stable HEK293T cells carrying inducible WT or MUT VMA21 were induced with doxycycline and loaded with the pH indicator dextran-conjugated LysoSensor Blue/Yellow. Untransfected cells were treated with EMS buffer calibrated to pH 3.5, pH 4.5 or pH 5.5. The fluorescence intensity of cell suspensions was read using flow cytometry. The mean fluorescence intensity of the yellow dye signal is a measure of lysosomal pH. (B) Estimation of lysosomal pH from data generated as in panel A with n = 3. (C) Vacuolar pH was measured using a pH-sensitive Pho8-SEP protein that exhibits stronger fluorescence with higher pH. Upper images show the fluorescence signal of Pho8-SEP; lower images show the corresponding light microscopy. (D) The quantitative analysis of fluorescence intensity of Pho8-SEP in WT, Vma21[∆66-77], and vma21∆ cells. Mean ± SD of n = 3 independent experiments. Unpaired, 2-tailed t-test with Bonferroni correction; *: p < 0.05, ***: p < 0.005. (E) Lysosomal V-ATPase activity (see Materials and Methods) in lysosomal preparations isolated via lyso-IP from cells carrying no lyso-IP bait (negative control), (-) empty vector, VMA21 WT, VMA21 93X, an shRNA targeting VMA21 or bafilomycin A1. n = 4, unpaired, 2-tailed t-test; ***: p < 0.005. (F) Growth of yeast strains (WT, Vma21[∆66-77], and vma21∆) on YPD plates buffered to pH 5.0 and on YPD plates buffered to pH 7.5. (G) The quantitative V-ATPase activity of WT, Vma21[∆66-77], and vma21∆ vacuoles were measured. After vacuole isolation, the release of inorganic phosphate arising from hydrolysis of ATP was determined. Mean ± SD of n = 3 independent experiments. Unpaired, 2-tailed t-test; ***: p < 0.005.
![Figure 6. Follicular-lymphoma associated VMA21 mutations reduce the ability of the V-ATPase to acidify lysosomes because of reduced lysosomal V-ATPase activity. (A) Stable HEK293T cells carrying inducible WT or MUT VMA21 were induced with doxycycline and loaded with the pH indicator dextran-conjugated LysoSensor Blue/Yellow. Untransfected cells were treated with EMS buffer calibrated to pH 3.5, pH 4.5 or pH 5.5. The fluorescence intensity of cell suspensions was read using flow cytometry. The mean fluorescence intensity of the yellow dye signal is a measure of lysosomal pH. (B) Estimation of lysosomal pH from data generated as in panel A with n = 3. (C) Vacuolar pH was measured using a pH-sensitive Pho8-SEP protein that exhibits stronger fluorescence with higher pH. Upper images show the fluorescence signal of Pho8-SEP; lower images show the corresponding light microscopy. (D) The quantitative analysis of fluorescence intensity of Pho8-SEP in WT, Vma21[∆66-77], and vma21∆ cells. Mean ± SD of n = 3 independent experiments. Unpaired, 2-tailed t-test with Bonferroni correction; *: p < 0.05, ***: p < 0.005. (E) Lysosomal V-ATPase activity (see Materials and Methods) in lysosomal preparations isolated via lyso-IP from cells carrying no lyso-IP bait (negative control), (-) empty vector, VMA21 WT, VMA21 93X, an shRNA targeting VMA21 or bafilomycin A1. n = 4, unpaired, 2-tailed t-test; ***: p < 0.005. (F) Growth of yeast strains (WT, Vma21[∆66-77], and vma21∆) on YPD plates buffered to pH 5.0 and on YPD plates buffered to pH 7.5. (G) The quantitative V-ATPase activity of WT, Vma21[∆66-77], and vma21∆ vacuoles were measured. After vacuole isolation, the release of inorganic phosphate arising from hydrolysis of ATP was determined. Mean ± SD of n = 3 independent experiments. Unpaired, 2-tailed t-test; ***: p < 0.005.](/cms/asset/02da538d-26e7-4643-9ba9-a703510090ce/kaup_a_2050663_f0006_oc.jpg)
We proceeded with complimentary measurements of the ability of the V-ATPase to acidify the vacuolar lumen in WT or mutant yeast cells. Pho8 was modified by fusing a pH-sensitive super ecliptic pHluorin (SEP) fluorophore protein on its C terminus; the fluorescent pH-biosenser Pho8-SEP displays higher fluorescence intensity at higher pH levels [Citation38]. The fluorescence intensity corresponding to Pho8-SEP was highest in vma21∆ mutant cells, with WT cells displaying the lowest signal (, D). The Vma21[∆66-77] mutant displayed an intermediate signal indicating an increased vacuolar pH compared with WT cells (, D).
The assembly of V-ATPase holoenzyme on lysosomes is preserved in VMA21 MUT cells but the activity of V-ATPase is reduced
Next, we measured V-ATPase activity in lysosomes isolated via lyso-IP from cells carrying WT or MUT VMA21. We also measured V-ATPase activity is cells treated with the V-ATPase inhibitor bafilomycin A1 as well as in cells treated with an shRNA targeting VMA21. We found that the activity of V-ATPase in lysosomes of VMA21 MUT cells treated with bafilomycin A1 was fully inhibited and that targeting VMA21 with an shRNA also substantially lowered V-ATPase activity. The V-ATPase activity in the VMA21 MUT was ~50% of WT providing a mechanistic explanation for the observed higher lysosomal pH ().
To determine whether the elevated vacuolar pH in Vma21[∆66-77] mutant yeast cells is caused by compromised V-ATPase activity, we first examined the growth phenotypes of WT and Vma21[∆66-77] mutant cells on plates buffered to pH 7.5. Yeast mutants lacking V-ATPase activity exhibit a distinctive set of growth defects including reduced growth at elevated pH [Citation39]. The Vma21[∆66-77] mutant showed a diminished ability to grow at pH 7.5 relative to the WT, and, in agreement with the intermediate vacuolar pH as assessed with Pho8-SEP, its phenotype was not as severe as that seen with the vma21 null mutant ().
We also directly measured V-ATPase activity in isolated vacuoles through detecting the hydrolysis of ATP in a bafilomycin A1-sensitive manner. Consistent with the growth phenotypes, the Vma21[∆66-77] mutant showed reduced V-ATPase activity equivalent to approximately 41% of the WT strain, whereas the V-ATPase activity dropped to approximately 9% of the WT in vma21 null cells ().
The aberrant metabolome of VMA21 MUT lysosomes defined via lyso-IP and lysosomal metabolomics
To identify the lysosomal metabolome changes imparted by MUT VMA21 on cells, we measured the relative amounts of small molecules in lysosomes isolated from cells carrying MUT or WT VMA21 or parental cells treated with bafilomycin A1. We isolated lysosomes with lyso-IP using limiting amounts of HA-beads, to precipitate equal amounts of intact lysosomes and subjected the lysosomes to untargeted small molecule metabolomics (Tables S3-4).
As an initial QC step, we correlated the relative amounts of metabolites in bafilomycin A1-treated cells with published data by Abu-Remaileh et al. [Citation35], using similar conditions and detected a high statistical correlation (R = 0.82). Next, we performed correlation analyses on bafilomycin A1-treated samples compared with VMA21 MUT samples and detected a high correlation (R = 0.89) for proteinogenic amino acids confirming a similarity in the lysosomal metabolome of cells with elevated lysosomal pH ().
Figure 7. Untargeted metabolomic analyses of lysosomes and the cytosolic fraction from cells expressing VMA21 WT or MUT. Intact lysosomes were isolated from HEK293T cells stably expressing the protein bait 3xHA-TMEM192 and inducible VMA21 WT and MUT using the lyso-IP method [Citation35]. An aliquot of cells was treated with bafilomycin A1 for 1 h and processed in parallel. The lysosomes and the post-IP supernatant were processed for untargeted metabolomics using tandem mass spectrometry. (A) LEFT: Pearson correlation of lysosomal amino acid ratios. X-axis: log2 MUT:WT and Y-axis: log2 BAF:WT. RIGHT: X-axis: log2 BAF:WT from this study and Y-axis: log2 BAF:WT from the study by Abu-Remaileh et al., Science 2017. (B) Volcano plot display for lysosomal di- and tripeptides. Left. X-axis: log2-fold ratios of di- and tripeptides in cells carrying MUT:WT VMA21. Y-axis: log10 FDR MUT:WT VMA21. Right. Data for bafilomycin A1-treated cells compared with VMA21 WT cells. (C) Volcano plot display for lysosomal amino acids. LEFT: X-axis log2-fold ratios of amino acids in cells carrying MUT:WT VMA21. Y-axis: log10 FDR. RIGHT: Data for bafilomycin A1-treated cells compared with VMA21 WT cells. (D) Volcano plot display for amino acids in the post-IP supernatant. LEFT: X-axis log2-fold ratios of amino acids in cells carrying MUT:WT VMA21. Y-axis: log10 FDR. RIGHT: Data for bafilomycin A1-treated cells compared with VMA21 WT cells. (E) Heatmap and numerical display of identified amino acids mass ratios for the indicated comparisons on top. Empty cells indicates the amino acid was not identified unequivocally for that condition. The mean results from three biological replicates are shown.
![Figure 7. Untargeted metabolomic analyses of lysosomes and the cytosolic fraction from cells expressing VMA21 WT or MUT. Intact lysosomes were isolated from HEK293T cells stably expressing the protein bait 3xHA-TMEM192 and inducible VMA21 WT and MUT using the lyso-IP method [Citation35]. An aliquot of cells was treated with bafilomycin A1 for 1 h and processed in parallel. The lysosomes and the post-IP supernatant were processed for untargeted metabolomics using tandem mass spectrometry. (A) LEFT: Pearson correlation of lysosomal amino acid ratios. X-axis: log2 MUT:WT and Y-axis: log2 BAF:WT. RIGHT: X-axis: log2 BAF:WT from this study and Y-axis: log2 BAF:WT from the study by Abu-Remaileh et al., Science 2017. (B) Volcano plot display for lysosomal di- and tripeptides. Left. X-axis: log2-fold ratios of di- and tripeptides in cells carrying MUT:WT VMA21. Y-axis: log10 FDR MUT:WT VMA21. Right. Data for bafilomycin A1-treated cells compared with VMA21 WT cells. (C) Volcano plot display for lysosomal amino acids. LEFT: X-axis log2-fold ratios of amino acids in cells carrying MUT:WT VMA21. Y-axis: log10 FDR. RIGHT: Data for bafilomycin A1-treated cells compared with VMA21 WT cells. (D) Volcano plot display for amino acids in the post-IP supernatant. LEFT: X-axis log2-fold ratios of amino acids in cells carrying MUT:WT VMA21. Y-axis: log10 FDR. RIGHT: Data for bafilomycin A1-treated cells compared with VMA21 WT cells. (E) Heatmap and numerical display of identified amino acids mass ratios for the indicated comparisons on top. Empty cells indicates the amino acid was not identified unequivocally for that condition. The mean results from three biological replicates are shown.](/cms/asset/060ff53e-fdaf-4295-9aa0-273ce490206e/kaup_a_2050663_f0007_oc.jpg)
Subsequently, we focused on di/tripeptides. We found that a variety of lysosomal di/tripeptides were much reduced in lysosomes isolated from VMA21 MUT cells compared with WT cells, suggesting decreased generation due to impaired proteolysis by pH-dependent hydrolases and peptidases (). Surprisingly, however, we found that the lysosomal concentrations of proteinogenic amino acids was not uniformly lower in VMA21 MUT lysosomes but instead comprised amino acids that were lower (e.g., histidine, asparagine, lysine, methionine, tryptophan) or higher (e.g., proline, alanine, glutamic acid, serine) than in VMA21 WT cells (, E). These data strongly suggested that the lysosomal amino acid concentration is determined not only by the rate of protein and peptide hydrolysis but also by pH-dependent changes in lysosomal amino acid handling and transport into the cytosol.
We also measured the relative amounts of amino acids in the post IP supernatant fraction and found that most amino acids were modestly lower in MUT than in WT cells (, E and Tables S5-6). The lowered cytoplasmic amino acid concentration likely activates the signal that increases autophagic flux in VMA21 MUT cells.
Primary human follicular lymphoma B cells carrying mutant VMA21 are sensitive to inhibition of autophagic flux
A recent report described the discovery of the novel potent ULK1 kinase inhibitor MRT68921 [Citation33,Citation40], and we had previously confirmed the ability of MRT68921 to inhibit the phosphorylation of the direct ULK1 substrate ATG13 [Citation18]. Here, we treated primary purified human FL B cells isolated from lymph node biopsies that carried WT or MUT VMA21 with escalating doses of MRT68921 and measured the cell fraction alive after 72 h using CellTiter-glow®-based viability. Given the preciousness of the FL biopsy material we combined the new FL WT data (N = 4; ; black curves) with data from ten similarly treated WT FL cases (; gray curves) previously published, as findings were very similar [Citation18]. We found that FL B cells carrying MUT VMA21 were substantially more sensitive to ULK1-inhibitor-induced cell death than VMA21 WT lymphoma cells which were not sensitive (IC50 values calculated based on the Hill equation of 1 µM and 4.5 µM, respectively) (; red curves. L34 was a bone marrow aspirate containing a mixture of FL B cells and normal B cells). Of additional interest, the two FL cases carrying the VMA21 p.93X mutation were most sensitive to MRT68921 treatment. We also reviewed our published findings on MUT ATP6V1B2 (; blue curves), which showed that all V-ATPase-impaired FL are sensitive to ULK1 inhibition. Finally, treatment of VMA21 MUT and WT FL B cells with SAR405 (a PIK3C3/VPS34 inhibitor) also showed heightened sensitivity of VMA21 MUT FL (Figure S4) [Citation41].
Figure 8. Follicular lymphoma-associated VMA21 mutations and dependence on autophagic flux for survival of primary FL B cells. (A) Primary human purified FL B cells carrying WT or mutant VMA21 were cultured in serum-supplemented RPMI 1640 medium containing 100% Leu and treated with the ULK1 inhibitor MRT68921 for 72 h at the indicated concentrations. Cell viability was measured using Celltiter-Glo® and is displayed normalized to the viability of untreated cells cultured for 72 h. Red: mutant VMA21; Black: WT VMA21. Gray: WT VMA21 (previously reported in [Citation18]). X-axis log10 scale. (B) Summary data calculated using the Hill equation. Standard deviations are plotted. IC50 WT 4.5 µM; IC50 MUT 1.0 µM. (C) Same data as in (B) plus, the addition of previously published data on ATP6V1B2 MUT FL [18] for comparison purposes only.
![Figure 8. Follicular lymphoma-associated VMA21 mutations and dependence on autophagic flux for survival of primary FL B cells. (A) Primary human purified FL B cells carrying WT or mutant VMA21 were cultured in serum-supplemented RPMI 1640 medium containing 100% Leu and treated with the ULK1 inhibitor MRT68921 for 72 h at the indicated concentrations. Cell viability was measured using Celltiter-Glo® and is displayed normalized to the viability of untreated cells cultured for 72 h. Red: mutant VMA21; Black: WT VMA21. Gray: WT VMA21 (previously reported in [Citation18]). X-axis log10 scale. (B) Summary data calculated using the Hill equation. Standard deviations are plotted. IC50 WT 4.5 µM; IC50 MUT 1.0 µM. (C) Same data as in (B) plus, the addition of previously published data on ATP6V1B2 MUT FL [18] for comparison purposes only.](/cms/asset/7c166e32-b0c5-493f-a5b3-7b133160f234/kaup_a_2050663_f0008_oc.jpg)
In summary, these important findings based on primary human FL B cells uncover a survival addiction to activated autophagic flux in VMA21 MUT FL B cells, providing support for novel therapeutic approaches targeting autophagy in FL.
High-throughput chemical screens identify highly potent inhibitors of autophagy induced by MUT VMA21
We conducted a high-throughput microscopy-based screen of a compound library containing 4,577 clinically approved drugs or tool compounds that could be repurposed, aiming at identification of small molecules that inhibited VMA21-MUT induced autophagy (Table S7). The rationale for these screens were two-fold: i) to further elucidate mechanisms of VMA21-MUT induced autophagy, and ii) to identify pathways and molecular targets suitable for therapeutic development in FL patients. We seeded HEK293T reporter cells carrying GFP-LC3-RFP fusion proteins into 384-well plates and left them untreated or induced VMA21 MUT protein with doxycycline and treated cells with experimental compounds at 4 µM. As controls we chose the ULK1 inhibitor MRT68921 and the PIK3C3/VPS34 inhibitor SAR405. To activate autophagy, we used torin 1.
The accumulation of GFP-stained green puncta following autophagy activation was quantitatively scored via high-throughput microscopy () and the percent inhibition of green puncta formation by compounds calculated from normalization against untreated cells (Tables S8-9). As can be seen in , the ULK1 inhibitor MRT68921 (black dots) resulted in ~90% inhibition across many replicates, while untreated cells (in blue) showed no inhibition. Many compounds (gray) were inactive while some in fact stimulated autophagy. We selected 112 primary screens hits (red), which demonstrated autophagy inhibition in the primary screen between 50% to > 95%, for triplicate assay repeats, and based on these results selected 32 compounds for further study ().
Figure 9. Results from a high-throughput chemical screen for compounds inhibiting VMA21-MUT induced autophagy. (A) UPPER: Images of HEK293T cells stably expressing the autophagy reporter fusion protein GFP-LC3-(ATG4 cleavage site)-RFP and inducible VMA21 MUT protein (p.93X). High-power inserts show the accumulation of GFP+ autophagosomes (APs) and autolysosomes (ALs). LOWER: Transformed image showing detection and quantification of APs and ALs. (B) Screening flow schema. (C) Overview of assay results for the entire compound library. Displayed is the percentage of cells with > 5 GFP puncta (APs + ALs) in compound-treated cells divided by untreated cells. Individual assay results for cell expression of VMA21 MUT as follows: Blue dots, treated with DMSO only (assay baseline); black dots, treated with the autophagy inhibitor MRT68921 targeting ULK1 (positive control); red dots, treated with various compounds showing autophagy inhibiting properties (primary screen hits); gray dots, treated with various inactive compounds. (D) Pathway enrichment and target analysis for compounds with autophagy inhibitory properties. (E) Dose-response curve of selected autophagy inhibiting compounds including MRT68921 (targeting ULK1) and SAR405 (targeting PIK3C3/VPS34), CDK inhibitors and homoharringtonin
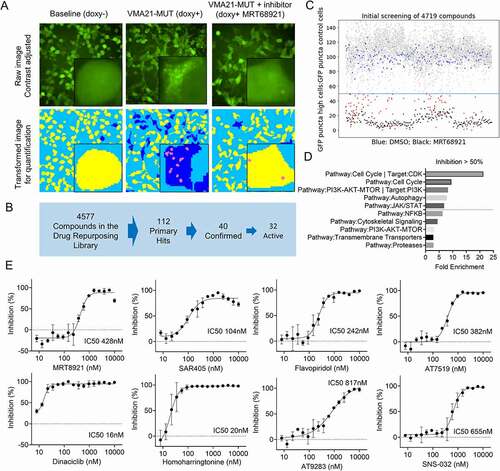
The review of these autophagy inhibitory compounds and their molecular targets gave interesting results. Multiple autophagy inhibitory drugs belonged to the class of cyclin-dependent kinase inhibitors, which hitherto had not received dedicated attention as autophagy inhibitors [Citation42]. A subsequent enrichment analysis comparing the fraction of autophagy inhibitory compounds within compound classes (e.g., CDK inhibitors or PIK3C3/VPS34 inhibitors) uncovered strong enrichment for CDK targets, autophagy components, phosphoinositide 3-kinase, and a few other pathways ().
Next, we derived dose-response curves for each of the 32 compounds ( and Figure S3). In these assays, MRT68921 displayed an IC50 of 428 nM and SAR405 of 104 nM. However, neither one of these compounds is in active clinical development. The clinical grade multi-targeted CDK inhibitor dinaciclib potently inhibited VMA21-MUT induced autophagy at an IC50 of 16 nM. We identified multiple other CDK inhibitors with potent autophagy-inhibiting properties including SNS-032, AT7519, AT9283, PHA-793887, AZD5438, R547, PHA-767491, abemaciclib, and LY2857785 ( and Figure S3). These compounds variably target CDKs 1, 2, 3, 5, 7, 8, and 9 with most of them targeting a CDK core set including CDK2, CDK7 and CDK9. Additional CDKs of interest that are targeted by these compounds are CDK16, CDK17 and CDK18 (PCTAIRE kinase family), which have recently been implicated in autophagy regulation [Citation43]. The clinically used drug ribociclib, targeting CDK4 and CDK6 was not active and the specific CDK7 inhibitor THZ1 was also only modestly active. Furthermore, RO-3306 with relative specificity for CDK1 and CDK2 was inactive. Together, these results narrow the CDK profile of interest to CDK9 and CDK16 to CDK18 but relevant targeting of other CDKs or non-CDKs remains possible.
Multiple other compounds with attractive properties, including the clinically used drugs flavopiridol (a pan CDK inhibitor; IC50 = 242 nM) and homoharringtonine (a ribosome and protein synthesis inhibitor; IC50 = 20 nM) approved for CML treatment were identified and confirmed ().
Next, we treated primary purified human FL WT or MUT for VMA21 as above with dinaciclib, flavopiridol or homoharringtonine (Figure S4). We found that all three drugs killed FL WT or MUT for VMA21 at low concentrations and thus could be considered for future FL clinical trials.
Discussion
In this study, we report on the identification of common mutations in VMA21, a small ER-resident chaperone protein for the vacuolar-type H+-translocating ATPase (V-ATPase) in 12% of follicular lymphoma (FL). The subsequent in-depth functional characterization uncovered that the FL-associated VMA21 hotspot nonsense mutation p.R93X, comprising 40% of all VMA21 mutations as well as p.88X and p.95X result in the deletion of a C-terminal non-canonical ER retrieval sequence, which causes mutant VMA21 protein to mis-localize to lysosomes. Consequently, there is a reduction in total lysosomal V-ATPase activity and a partial inability to fully acidify the lysosomes. The pathological elevation in lysosomal pH caused a defect in the degradation of lysosomal cargo proteins as measured through lysosomal metabolomics and a reduction in the concentration of cytosolic amino acids. Overall, this aberrant lysosomal phenotype associated with a substantial compensatory increase in autophagic flux, which we here demonstrate constitutes an Achilles heel survival dependency for FL. Targeting the apical autophagy regulating ULK1 kinase complex with a small molecule inhibitor resulted in the death of VMA21 MUT FL B cells, while VMA21 WT cells were largely resistant to such treatment. Therefore, we are identifying the compensatory activation of autophagic flux as a survival dependency and its inhibition as a novel therapeutic opportunity in FL with reduced V-ATPase function.
FL harbors mutations in ATP6V1B2, VMA21, ATP6AP1 and ATP6AP2, and sporadic mutations in other V-ATPase proteins, which together affect one third of all FL. As such, the V-ATPase emerges as a commonly targeted enzyme in FL. In prior work, we have shown that mutations in the V-ATPase V1 subunit ATP6V1B2 create a catalytically hypomorphic V-ATPase that results in elevated lysosomal pH and activated autophagy and a dependency for survival. The work presented here unifies the phenotypes of FL with mutations in ATP6V1B2 and VMA21 and converges on a novel pathobiological mechanism driving FL. Last, how mutated ATP6AP1 and ATP6AP2 affect these processes and FL biology is unknown and awaits ongoing detailed study.
The recently reported cryo-EM structures of V-ATPase holoenzymes purified from brain and other sources placed ATP6AP1 and ATP6AP2 proteins centrally within the membrane-spanning ring-like structures formed by nine V-ATPase ATP6V0C subunits and both proteins were identified as central components of the mature V-ATPase. Of interest, the VMA21 protein was absent in all reported structures implying that it interacts only transiently with the V-ATPase holoenzyme or was lost during enzyme purification. In the VMA21 IP-mass spec experiments reported here we detected the entire V-ATPase holoenzyme, including V0, V1 and ATP6AP1-ATP6AP2 in both VMA21 WT and MUT immunoprecipitates but measured increased amounts of co-immunoprecipitating proteins with the mutants. The increased co-immunoprecipitation of MUT VMA21 and V-ATPase holoenzyme together with results from confocal microscopy studies supports the idea that MUT VMA21 remains aberrantly associated with V-ATPase in the lysosomes. This aberrant association results in only a partially functional or possibly immature V-ATPase enzyme or in the direct inhibition of V-ATPase activity by aberrantly associated MUT VMA21 protein.
We previously demonstrated that purified human primary FL B cells carrying MUT ATP6V1B2 acquire the ability to survive in lowered or no leucine-containing media. Considering the lowered cytoplasmic leucine and amino acid concentration that we measured in VMA21 MUT cells together with these findings seem paradoxical at first. Leucine signals to MTORC1 via different mechanisms including its metabolite acetyl-CoA [Citation44,Citation45], the latter being generated via multiple metabolic pathways. One possible explanation relies on metabolic adaptation by MUT FL B cells allowing these cells to preferentially use alternative fuel sources to generate acetyl-CoA and, in this context, it is relevant that we found that dichloroacetate/DCA, known to elevate intracellular acetyl-CoA levels, abolishes aberrant activation of autophagy in VMA21 MUT cells [Citation46].
In addition to resistance to nutrient starvation as likely exists in pathologically enlarged lymph nodes, why do FL frequently select for mutations that cause lysosomal dysfunction and compensatory elevations in autophagy flux? Considering the important role of the lysosome in MHC peptide loading and B cell antigen presentation, is the elevated lysosomal pH permitting immunological escape from T cells? Is the elevated autophagy needed to overcome impairments to baseline autophagy as part of the high BCL2 protein level in FL and the effects on the BCL2-BECN1-PIK3C3/VPS34 complexes [Citation47], and could BCL2 inhibitors be used in combination with autophagy inhibitors as therapy for FL?
To exploit novel therapeutic opportunities derived from the survival addiction of VMA21 MUT FL on intact autophagic flux, we conducted a screen for compounds that could inhibit MUT-VMA21 induced autophagy. Through these efforts we identified several cyclin-dependent kinase inhibitors (e.g., dinaciclib, flavopiridol, and multiple others) that strongly abolished autophagy activation and demonstrated their ability to kill purified FL cells at low concentrations. Of course, these drugs have other known effects, which would need to be considered when planning further study and possible development as FL therapeutics. These results also motivate future studies into the role of specific CDKs in autophagy regulation in lymphoma, and recent findings of involvement of CDK16 in autophagy activation could serve as a mechanistic lead [Citation43].
In summary, our studies elucidate novel mechanisms of action of FL-associated mutations in chaperones and components of the V-ATPase and provide a framework for future biological or therapeutic studies of hypomorphic or hypofunctional V-ATPase in FL. Our findings open up novel avenues for the treatment of FL, possibly through intermittent inhibition of autophagy [Citation41,Citation48], and we identify multiple hitherto unsuspected compounds as attractive in this setting [Citation33,Citation40,Citation49].
Materials and Methods
Patient characteristics and study approvals
Results from the Sanger-based resequencing of exons 1–3 and adjacent intronic sequences of VMA21 using template genomic DNA isolated from 151 flow-sorted FL and 19 transformed FL (t-FL) are reported here [Citation50]. Lymphoma patients were enrolled into two lymphoma repositories at the University of Michigan Comprehensive Cancer Center (IRBMED #HUM00007985 and IRBMED #HUM00017055) after signing informed consent documents approved by the University of Michigan Institutional Review Board (IRBMED). All investigations were performed in accordance with ethical guidelines outlined in the declaration of Helsinki. Genomic research on all specimens was approved through IRBMED #HUM00005467. We furthermore analyzed by Sanger sequencing DNA from 49 flow-sorted FL cases collected between 1990 and 2005 as previously described that were derived from de-identified leftover clinical material [Citation51].
Exon resequencing of VMA21 in FL
Primers to amplify and sequence VMA21 were designed using the primer 3 program (https://bioinfo.ut.ee/primer3-0.4.0/) and sequences generated using direct sequencing. The sequence traces were analyzed using Mutation Surveyor® software (Softgenetics®) and by visual inspection. Mutations were confirmed to be somatically acquired using unamplified lymphoma-cell derived DNA and paired CD3-cell derived DNA isolated from flow-sorted LN or blood T cells or from buccal DNA.
VMA21 cDNA mutagenesis and lentiviral vector generation, cell transfection and cell transduction to generate stable cell lines
Cell lines: The OCI-LY1 lymphoma cell line was obtained from the Ontario Cancer Institute, Toronto, Canada. Cell line authentication was performed through sequence analysis of known gene mutations. The HEK293T cell line was purchased from Takara (632180).
Reagents and mutagenesis
A pENTER-CMV-plasmid containing the VMA21 cDNA NM_001017980 was purchased from Vigene Biosciences (CH893482) and was used to generate MUT VMA21 cDNAs using the QuikChange® Lightning Site-Directed Mutagenesis Kit (Stratagene/Agilent, 210518). Full-length WT and MUT N-terminal HA-tagged VMA21 were constructed using PCR and cloned into the doxycycline-inducible plasmid pCW57.1 (Addgene, 41393; deposited by Dr. David Root) or pCW57-MCS1-P2A-MCS2 (pCW57-blast; Addgene, 80921; deposited by Dr. Adam Karpf) and sequence verified.
Generation of stable doxycycline-inducible lymphoma cell and 293T cell lines
The methods followed our previously published detailed protocols [Citation18] and cell pools were used for experiments described herein.
Autophagy assays in cell lines inducibly expressing VMA21 WT or MUT
Measurement of LC3-II in VMA21-inducible cell lines
Doxycycline-inducible 293 T cell lines expressing WT or MUT VMA21 were cultured in full DMEM medium (Gibco, 2311544) supplemented with 10% FBS and induced with 1 µg/ml doxycycline (Cayman Chemical, 14422) for 48 h followed by a 4-h washout period, a condition used throughout this study. Aliquots of cells were treated with the V-ATPase inhibitor bafilomycin A1 (Cayman Chemical, 11038) at 100 nM for 4 h. The cell pellets were lysed in lysis buffer (LB) containing 1% NP-40 (Sigma, 18896), 150 mmol/l NaCl, 25 mmol/l Tris, pH 8.0, 20 mmol/l NaF, 2 mmol/l EGTA, 2 mmol/l EDTA, supplemented with protease inhibitors (Sigma-Aldrich, P3840), phosphatase inhibitors (Sigma-Aldrich, P0044), sodium orthovanadate (Sigma-Aldrich, 450243), and PMSF (Thermo Fisher Scientific, 36978). The detergent-soluble fraction of cell lysates was cleared by centrifugation at 17,000 g for 10 min. Protein was fractionated through SDS-PAGE and prepared for immunoblotting. Antibodies used for immunoblotting are listed in . Band intensities were analyzed by densitometry (ImageJ, version 2.0.0, NIH).
Table 1. Antibodies and antisera used in this study.
Measurement of autophagic flux using electron microscopy
Stable HEK293T cell lines overexpressing WT or MUT HA-VMA21 were induced with doxycycline as above. Cells were then fixed with 2% glutaraldehyde (EM grade) in 0.2 M HEPES, pH 7.4 at room temperature for 2 h. During fixation, the cells were scraped off the culture dish and pelleted. The fixed cell pellets were kept at +4°C until sample preparation for transmission electron microscopy. Images were taken at 3000 x primary magnification, using the principle of uniform random sampling, and analyzed using ImageJ. Each image was zoomed in on the screen for counting of profiles. The cytoplasmic area was estimated using point counting. The number of autophagosomes, autolysosomes and endolysosomes was counted for ~70 to 90 images from each group.
Autophagy assays using the GFP-LC3-RFP reporter in VMA21-inducible cell lines
Generation of GFP-LC3-RFP reporter cell lines
HEK293T cells were transfected with pMRX-IP-GFP-LC3-RFP (Addgene, 84573; deposited by Dr. Noboru Mizushima) and packaging plasmids and virus-containing supernatant media collected 48 h after transfection. The virus-containing media were used to transduce HEK293T cells and OCI-LY1 lymphoma cells. After viral transduction, cells were treated with 2 µg/ml of puromycin for 2 days. After 3–5 days of culture in full medium, cells were than infected with pCW57-blast lentiviruses carrying WT or MUT HA-VMA21 cDNAs followed by selection with 5 µg/ml blasticidin S (Cayman Chemical, 14499). The expression of GFP-LC3-RFP was verified by fluorescence microscopy and immunoblotting and the inducible expression of VMA21 was verified by immunoblotting.
Measurement of GFP-positive autophagosomes and autolysosomes
Doxycycline-inducible stable 293T expressing GFP-LC3-RFP and VMA21 WT or MUT cell lines were cultured on chambered cover glasses and induced with doxycycline as above. Cell Images were taken using a Leica SP5 confocal imaging system.
easurement of free GFP
Doxycycline-inducible stable 293T cells or OCI-LY1 cells expressing GFP-LC3-RFP and VMA21 WT or MUT were cultured in DMEM medium supplemented with 10% FBS and induced with doxycycline as above. Cells were harvested and prepared for immunoblotting. Band intensities were analyzed by densitometry (ImageJ, version 2.0.0, NIH).
Intracellular localization of WT and MUT HA-VMA21 in HEK293T cells using confocal microscopy
Cells were plated on pre-sterilized chambered cover glasses. The expression of HA-VMA21 WT and MUT was induced with doxycycline as above. Cells were fixed and permeabilized with 4% paraformaldehyde for 10 min at room temperature (RT) and subsequently in 0.1% Triton X-100 (Sigma, T8532) for 5 min at 4°C. Samples were blocked with 4% bovine serum albumin (BSA; Dot Scientific, DSA30075-100) in phosphate-buffered saline (PBS; Lonza, 17–516 F) for 1 h at RT. Staining was performed using primary antibodies () diluted in 4% BSA in PBS. Species-specific conjugated secondary antibodies were diluted 1:1000 in 4% BSA in PBS and applied for 1 h at RT. Nuclei were counterstained with 4′,6-diamidino-2-phenylindole (DAPI; Molecular Probes/Fisher, D1306). The coverslips were mounted onto glass slides with ProLong® Gold Antifade Mountant reagent (Thermo Fisher, P36930). Images were taken using a Leica SP5 confocal imaging system. Pearson’s correlation coefficient analysis was used to determine colocalization and was performed using ImageJ v2.0.0 software.
Measurement of V-ATPase components and metabolites in isolated lysosomes
Cell line generation and lysosome isolation
The lysosome isolation was done using the rapid Lyso-IP method developed by Abu-Remaileh et al, 2017 [Citation35]. Briefly, HEK293T cells stably expressing the lysosomal bait protein 3xHA-TMEM192 were generated using the lentiviral plasmid pLJC5-Tmem192-3xHA (Addgene, 102930; deposited by Dr. David Sabatini) as above. The control HEK293T cells were generated expressing 2xFlag-TMEM192 using pLJC5-Tmem192-2xFlag (Addgene, 102929; deposited by Dr. David Sabatini). After puromycin selection the HEK293T cells were transduced with pCW57(blast) lentiviruses carrying cDNA encoding non-tagged WT or MUT VMA21, respectively, followed by selection using 5 µg/ml blasticidin S. To perform isolation of intact lysosomes via immunoprecipitation (Lyso-IP), cells were lysed using a Dounce homogenizer and the lysates were centrifuged at 1000 x g for 2 min at 4°C. The supernatant containing the cellular organelles including lysosomes was subsequently immunoprecipitated using anti-HA-conjugated magnetic beads (Thermo Scientific, 88836), followed by three quick washings using ice cold Tris-buffered saline (TBS; Bio-Rad, 1706435). After immunopurification, the bead-bound lysosomes were snap frozen in liquid nitrogen.
Measurement of V-ATPase components in lysosomes
The 3xHA-TMEM192-expressing 293T cells carrying inducible VMA21 WT or MUT were induced with doxycycline as above. Lysosomes were isolated from 1.5 × 107 cells and using a limiting amount (25 µL) of anti-HA beads and the bead-bound lysosomes subsequently lysed using standard lysis buffer for immunoblotting as described above. In parallel, 106 cells were harvested and lysed directly using the same buffer to obtain whole-cell lysates. Immunoblottings for various V-ATPase components were performed on protein from lysosomal and whole cell lysates.
Untargeted lysosomal and cytoplasmic metabolomics assay
The 3xHA-TMEM192-expressing 293T cells (108 cells/construct and condition for lysosomal metabolomics and 107 cells for cytoplasmic metabolomics) carrying inducible VMA21 WT or MUT were induced with doxycycline as above. Lysosomes were isolated using anti-HA beads. For lysosomal metabolomics, the IP-enriched lysosomes and the post-IP supernatants were kept and snap frozen with liquid nitrogen and subsequently processed in the Metabolomics Core at the University of Michigan. Small molecules were extracted, and untargeted metabolomics profiling was conducted using an HPLC/Q-TOF Mass Spec system. Manual peak integration was combined with automated peak calling to identify metabolites. Data reduction was performed with the software Binner (https://doi.org/10.1093/ bioinformatics/btz798). The accurate mass and retention time was then used for the database search against RefMet (MetabolomicsWorkbench.org). Features of interest from the database search were individually confirmed by tandem mass spectrometry (MS/MS) analysis.
Immunoprecipitation followed by quantitative tandem mass spectrometry
A total of 5 × 107 OCI-LY1 cells inducibly overexpressing HA-VMA21 WT or MUT were induced with doxycycline as above. Cells were pelleted, washed, and lysed on ice for 20 min in modified lysis buffer containing 0.2% NP-40 Surfact-Amps Detergent Solution (Thermo Fisher Scientific, 85124) as above. The detergent-soluble fraction of the cell lysates was obtained by centrifugation at 20,817 x g for 10 min. A total of 200 μL of protein A agarose beads (Cell Signaling Technology, 9863) were blocked with 20 μL total mouse IgG-UNLB (Southern Biotech, 0107–01) for 2 h. The blocked protein A agarose beads were washed and resuspended in lysis buffer. To preclear the protein samples, 50 μL conjugated protein A agarose beads were added to the protein lysate for 2 h at 4°C. Next, the anti–HA-loaded beads (Sigma-Aldrich, A2095) were blocked with 5% BSA in TBS, 0.05% Tween-20 (Sigma, P7949) TBST for 2 h, followed by washing with lysis buffer. A total of 40 μL of anti-HA-conjugated beads were added to precleared cell lysates and incubated with rotation for 5 h at 4°C. The beads were washed four times with lysis buffer containing 150 mmol/L NaCl and pelleted by centrifugation at 2,000 × g for 5 min.
The methods for protein SDS-PAGE and mass spectrometry using nano LC/MS-MS with a Waters M-Class high-performance liquid chromatography (HPLC) system interfaced to a Thermo Fisher Fusion Lumos mass spectrometer and data analysis using Mascot (Matrix Science) were as recently published [Citation52].
Measurement of lysosomal pH in HEK293T cells expressing WT or MUT HA-VMA21
HEK293T cells inducibly expressing WT or MUT HA-VMA21 were grown in 24-well plates and induced with doxycycline as above. A knockdown using lentiviruses carrying VMA21 shRNA (Millipore Sigma, TRCN0000122433) or scrambled shRNA was performed in parallel. LysoSensor Yellow/Blue dextran (Thermo Fisher Scientific, L22460) was added at 500 μg/mL and cells incubated for 16 h, followed by a 1-h chase in dextran/dye-free medium. The methods otherwise follow our published protocols [Citation18].
Measurements of lysosomal V-ATPase activity
HEK293T cells stably expressing TMEM192-3xHA or TMEM192-2xFlag and carrying inducible non-tagged VMA21 WT or MUT were induced with doxycycline as above. Lysosomes were subsequently isolated using Lyso-IP as described above. The V-ATPase activity was measured in the isolated lysosomes using the ATPase/GTPase Activity Assay Kit (Sigma-Aldrich, MAK113), and 4 mM ATP (Sigma-Aldrich, A7699). The absorbance was read at 620 nM using a plate reader. Cells treated with 100 nM bafilomycin A1 for 2 h were used as positive control for complete inhibition of V-ATPase.
High-throughput screening for compounds inhibiting MUT-VMA21 induced autophagy
The autophagy reporter 293T cells expressing GFP-LC3-RFP and inducible VMA21 WT or MUT were plated into black 384-well tissue culture plates (PerkinElmer, 6005182) at a density of 5000 cells/well using a multidrop device (Thermo Fisher Scientific, Waltham, MA). Phenol-red free DMEM (Gibco, 21063029) was used for cell culture. Doxycycline was added at a concentration of 1 µg/ml. The compound library used for drug screening (Drug Repurposing Set) was created by the Center for Chemical Genomics at the University of Michigan. The Drug Repurposing Set is a collection of FDA-approved compounds and selected clinical drug candidates (Table S7). The compounds were added using a liquid handler (Sciclone Advanced Liquid Handling 3000 V&P pintool) in 0.2-µL volume (final DMSO concentration = 0.4%). All compounds were tested as singletons at 4 µM. The ULK1 inhibitor MRT68921 (Cayman Chemical, 1190379) at 1 µM was added to one column of 16 wells on each plate as a control for autophagy inhibition (internal plate and assay control). Cells without doxycycline treatment (uninduced cells) were also plated into one column on each plate as an assay baseline control. Following the addition of doxycycline and compounds the plates were incubated for 24 h at 37°C in 5% CO2. The 384-wells plates were than imaged and analyzed using an ImageXpress Micro Widefield High Content Screening System at the Center for Chemical Genomics, Life Sciences Institute, University of Michigan. Next, the number of GFP-positive puncta per cell were quantified. Cells with more than or equal to five GFP-positive puncta were categorized as autophagy-activated cells and the total number of these cells were divided by the total number of diffusely GFP+ cells in the same high-power image. A hit was defined as a compound able to inhibit the number of MUT-VMA21 associated autophagy-activated cells to a level ≤50% of the doxycycline-induced VMA21-MUT expression control group. The average inhibition of MRT68921-positive controls was 86% with a Z’ value > 0.4. Hits from the primary screen and selected compounds targeting CDKs were further tested in secondary screenings at 4 µM in triplicates. Compounds validated in the secondary confirmatory screen were individually subjected to dose-response assays performed in duplicate at concentrations from 8.67 nM to 10 µM.
FL B cell column purification
Cryopreserved single-cell suspensions from human FL biopsy samples were thawed, washed and depleted of CD3+ T cells and CD14+ macrophages using Miltenyi beads and columns (Miltenyi, 130–050-101 and 130–050-201) following our published protocols [Citation18].
Drug treatment and cell viability assays of purified FL B cells
Purified FL B cells were plated in RPMI 1640 medium (Hyclone, SH30027.01) supplemented with 20% heat inactivated FBS at a density of 106 cells/ml and cultured at 37°C with 5% CO2. Torin 1 (10997) and SAR405 (16979) were procured from Cayman Chemical. Apilimod (SML2974) was procured from Millipore Sigma. Flavopiridol (S1230), pitavastatin (S1759), homoharringtonine (S9015) and dinaciclib (S2768) were purchased from Selleck Chemicals. Stock solutions of 100 mM were made with DSMO and were diluted serially in medium. DMSO-only was used as vehicle control. After 72 h of incubation, cell viability was measured with Celltiter-Glo® (Promega, G7572) and quantified on a microplate luminescence reader.
Yeast methods
Strains, media, and growth conditions
Yeast strains (SEY6210 and WLY176) were used to generate the Vma21[∆66-77] mutation and vma21∆ in the genome using standard methods [Citation53,Citation54]. Cells were cultured in rich medium (YPD; 1% [w:v] yeast extract, 2% [w:v] peptone], and 2% [w:v] glucose) as nutrient-rich conditions. Autophagy was induced through nitrogen starvation by shifting cells in mid-log phase from YPD to SD-N (0.17% yeast nitrogen base without ammonium sulfate or amino acids [ForMedium, CYN0501], and 2% [w:v] glucose) for the indicated times, referred to as starvation conditions. For growth assays, cells were grown in YPD, pH 5 to mid-log phase and then serially diluted and transferred to YPD plates buffered to pH 5 or pH 7.5. Growth phenotypes were then determined after 36 h at 30°C incubation.
Autophagy assays
Pho8∆60, GFP-Atg8, prApe1 processing, and Atg8 lipidation analyses were performed as previously described [Citation55–57].
Vacuole assays
Vacuole purification was performed as previously described [Citation58]. Vacuole lysates were prepared and subjected to SDS-PAGE. Vma2 and Vma4 were detected by western blot. Antisera to Vma2 [Citation59] was used at 1:5,000 dilution, and anti-Vma4 [Citation60] was used at 1:3,000 dilution. V-ATPase activity (ATP hydrolysis rate) was quantified colorimetrically as the phosphate released based on a previous method [Citation61]. Bafilomycin A1 inhibition was measured by adding bafilomycin A1 directly to the assay mixture to a final concentration of 100 nm. Specific V-ATPase activity represents the rate of bafilomycin A1-sensitive ATP hydrolysis.
Fluorescence Pho8-SEP assay
Vacuolar fluorescence of Pho8-SEP was monitored as previously described [Citation18].
Statistical Methods
Statistical comparisons between two conditions were carried out using unpaired two-sample t-tests or the Mann-Whitney U-test (where indicated). Bonferroni adjustments were used when a positive result from any one of several tests was used to support a single hypothesis [Citation62]. For analysis of metabolism data, unpaired two-sample t-tests and false discovery rate correction was calculated using Scipy and Statsmodels Python packages [Citation63,Citation64]. Volcano plots were drawn using the Matplotlib Python package [Citation65]. For concentration-response curves in chemical screening experiments and FL kill assays, sigmoidal dose–response models with variable slope for inhibition (based on the Hill equation) were used to estimate the IC50 using commercial software (GraphPad Prism). All bar plots, line plots and heatmaps were draw using GraphPad Prism.
Supplemental Material
Download Zip (35.2 MB)Acknowledgments
We are grateful for services provided by the genomics, metabolomics, proteomics, bioinformatics, and flow cytometry cores of the University of Michigan Rogel Comprehensive Cancer Center. We thank the Laboratory of Electron Microscopy, Institute of Biomedicine, University of Turku.
Disclosure statement
SNM owns shares in ABBVIE
Supplementary material
Supplemental data for this article can be accessed here
Additional information
Funding
Related Research Data
References
- Morton LM, Wang SS, Devesa SS, et al. Lymphoma incidence patterns by WHO subtype in the United States, 1992-2001. Blood. 2006 Jan 1;107(1):265–276.
- Casulo C, Byrtek M, Dawson KL, et al. Early relapse of follicular lymphoma after rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone defines patients at high risk for death: an analysis from the national lymphocare study. J Clin Oncol. 2015 Aug 10;33(23):2516–2522.
- Kridel R, Sehn LH, Gascoyne RD, et al. Pathogenesis of follicular lymphoma. J Clin Invest. 2012 Oct 1;122(10):3424–3431.
- Kahl BS, Yang DT. Follicular lymphoma: evolving therapeutic strategies. Blood. 2016 Apr 28;127(17):2055–2063.
- Kuppers R, Stevenson FK. Critical influences on the pathogenesis of follicular lymphoma. Blood. 2018 May 24;131(21):2297–2306.
- Okosun J, Wolfson RL, Wang J, et al. Recurrent mTORC1-activating RRAGC mutations in follicular lymphoma. Nat Genet. 2016 Feb;48(2):183–188.
- Ying ZX, Jin M, Peterson LF, et al. Recurrent Mutations in the MTOR Regulator RRAGC in follicular lymphoma. Clin Cancer Res. 2016 Nov 1;22(21):5383–5393.
- Oricchio E, Katanayeva N, Donaldson MC, et al. Genetic and epigenetic inactivation of SESTRIN1 controls mTORC1 and response to EZH2 inhibition in follicular lymphoma. Sci Transl Med. 2017;Jun 28;9(396): eaak9969.
- Bar-Peled L, Schweitzer LD, Zoncu R, et al. Ragulator is a GEF for the rag GTPases that signal amino acid levels to mTORC1. Cell. 2012 Sep 14;150(6):1196–1208.
- Sabatini DM. Twenty-five years of mTOR: uncovering the link from nutrients to growth. Proc Natl Acad Sci U S A. 2017 Nov 7;114(45):11818–11825.
- Kim E, Goraksha-Hicks P, Li L, et al. Regulation of TORC1 by Rag GTPases in nutrient response. Nat Cell Biol. 2008 Aug;10(8):935–945.
- Sancak Y, Peterson TR, Shaul YD, et al. The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science. 2008 Jun 13;320(5882):1496–1501.
- Wang S, Tsun ZY, Wolfson RL, et al. Metabolism. Lysosomal amino acid transporter SLC38A9 signals arginine sufficiency to mTORC1. Science. 2015 Jan 9;347(6218):188–194.
- Rebsamen M, Pochini L, Stasyk T, et al. SLC38A9 is a component of the lysosomal amino acid sensing machinery that controls mTORC1. Nature. 2015 Mar 26;519(7544):477–481.
- Jung J, Genau HM, Behrends C, et al. Amino acid-dependent mTORC1 regulation by the lysosomal membrane protein SLC38A9. Mol Cell Biol. 2015 Jul;35(14):2479–2494.
- Zoncu R, Bar-Peled L, Efeyan A, et al. mTORC1 senses lysosomal amino acids through an inside-out mechanism that requires the vacuolar H(+)-ATPase. Science. 2011 Nov 4;334(6056):678–683.
- Green MR, Kihira S, Liu CL, et al. Mutations in early follicular lymphoma progenitors are associated with suppressed antigen presentation. Proc Natl Acad Sci U S A. 2015 Mar 10;112(10):E1116–25.
- Wang F, Gatica D, Ying ZX, et al. Follicular lymphoma-associated mutations in vacuolar ATPase ATP6V1B2 activate autophagic flux and mTOR. J Clin Invest. 2019 Mar 4;129(4):1626–1640.
- Cotter K, Stransky L, McGuire C, et al. Recent Insights into the Structure, Regulation, and Function of the V-ATPases. Trends Biochem Sci. 2015 Oct;40(10):611–622.
- Wang R, Long T, Hassan A, et al. Cryo-EM structures of intact V-ATPase from bovine brain. Nat Commun. 2020 Aug 6;11(1):3921.
- Abbas YM, Wu D, Bueler SA, et al. Structure of V-ATPase from the mammalian brain. Science. 2020 Mar 13;367(6483):1240–1246.
- Ballabio A, Bonifacino JS. Lysosomes as dynamic regulators of cell and organismal homeostasis. Nat Rev Mol Cell Biol. 2020 Feb;21(2):101–118.
- Malkus P, Graham LA, Stevens TH, et al. Role of Vma21p in assembly and transport of the yeast vacuolar ATPase. Mol Biol Cell. 2004 Nov;15(11):5075–5091.
- Ryan M, Graham LA, Stevens TH, et al. Voa1p functions in V-ATPase assembly in the yeast endoplasmic reticulum. Mol Biol Cell. 2008 Dec;19(12):5131–5142.
- Guida MC, Hermle T, Graham LA, et al. ATP6AP2 functions as a V-ATPase assembly factor in the endoplasmic reticulum. Mol Biol Cell. 2018 Sep 1;29(18):2156–2164.
- Wang L, Wu D, Robinson CV, et al. Structures of a complete human V-ATPase reveal mechanisms of its assembly. Mol Cell. 2020 Nov 5;80(3):501–511 e3.
- Kabeya Y, Mizushima N, Ueno T, et al. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000 Nov 1;19(21):5720–5728.
- Nair U, Yen W-L, Mari M, et al. A role for Atg8–PE deconjugation in autophagosome biogenesis. Autophagy. 2012;8(5):780–793.
- Noda T, Klionsky DJ. The quantitative Pho8Delta60 assay of nonspecific autophagy. Methods Enzymol. 2008;451:33–42.
- Hutchins MU, Klionsky DJ. Vacuolar localization of oligomeric α-mannosidase requires the cytoplasm to vacuole targeting and autophagy pathway components in Saccharomyces cerevisiae. J Biol Chem. 2001;276(23):20491–20498.
- Kaizuka T, Morishita H, Hama Y, et al. An autophagic flux probe that releases an internal control. Mol Cell. 2016 Nov 17;64(4):835–849.
- Ronan B, Flamand O, Vescovi L, et al. A highly potent and selective Vps34 inhibitor alters vesicle trafficking and autophagy. Nat Chem Biol. 2014 Dec;10(12):1013–1019.
- Petherick KJ, Conway OJ, Mpamhanga C, et al. Pharmacological inhibition of ULK1 kinase blocks mammalian target of rapamycin (mTOR)-dependent autophagy. J Biol Chem. 2015 Nov 27;290(48):28726.
- Hill KJ, Stevens TH. Vma21p is a yeast membrane protein retained in the endoplasmic reticulum by a di-lysine motif and is required for the assembly of the vacuolar H(+)-ATPase complex. Mol Biol Cell. 1994 Sep;5(9):1039–1050.
- Abu-Remaileh M, Wyant GA, Kim C, et al. Lysosomal metabolomics reveals V-ATPase- and mTOR-dependent regulation of amino acid efflux from lysosomes. Science. 2017 Nov 10;358(6364):807–813.
- Sehnal D, Bittrich S, Deshpande M, Svobodová R, Berka K, Bazgier V, Velankar S, Burley S K, Koča J, Rose A S. (2021). Mol* Viewer: modern web app for 3D visualization and analysis of large biomolecular structures. Nucleic Acids Res, 49(W1), W431–W437. https://doi.org/10.1093/nar/gkab314
- Cannata Serio M, Graham LA, Ashikov A, et al. Mutations in the V-ATPase assembly factor VMA21 cause a congenital disorder of glycosylation with autophagic liver disease. Hepatology. 2020 Dec;72(6):1968–1986.
- Hughes AL, Gottschling DE. An early age increase in vacuolar pH limits mitochondrial function and lifespan in yeast. Nature. 2012 Dec 13;492(7428):261–265.
- Sambade M, Alba M, Smardon AM, et al. A genomic screen for yeast vacuolar membrane ATPase mutants. Genetics. 2005;170(4):1539–1551.
- Egan DF, Chun MG, Vamos M, et al. Small molecule inhibition of the autophagy kinase ULK1 and identification of ULK1 substrates. Mol Cell. 2015 Jul 16;59(2):285–297.
- Gayle S, Landrette S, Beeharry N, et al. Identification of apilimod as a first-in-class PIKfyve kinase inhibitor for treatment of B-cell non-Hodgkin lymphoma. Blood. 2017 Mar 30;129(13):1768–1778.
- Wells CI, Vasta JD, Corona CR, et al. Quantifying CDK inhibitor selectivity in live cells. Nat Commun. 2020 Jun 2;11(1):2743.
- Dohmen M, Krieg S, Agalaridis G, et al. AMPK-dependent activation of the Cyclin Y/CDK16 complex controls autophagy. Nat Commun. 2020 Feb 25;11(1):1032.
- Son SM, Park SJ, Lee H, et al. Leucine signals to mTORC1 via its metabolite acetyl-coenzyme A. Cell Metab. 2019 Jan 8;29(1):192–201 e7.
- He A, Chen X, Tan M, et al. Acetyl-CoA derived from hepatic peroxisomal beta-oxidation inhibits autophagy and promotes steatosis via mTORC1 activation. Mol Cell. 2020 Jul 2;79(1):30–42 e4.
- Marino G, Pietrocola F, Eisenberg T, et al. Regulation of autophagy by cytosolic acetyl-coenzyme A. Mol Cell. 2014 Mar 6;53(5):710–725.
- Pattingre S, Tassa A, Qu X, et al. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell. 2005 Sep 23;122(6):927–939.
- Rebecca VW, Nicastri MC, McLaughlin N, et al. A unified approach to targeting the lysosome’s degradative and growth signaling roles. Cancer Discov. 2017 Nov;7(11):1266–1283.
- Lazarus MB, Novotny CJ, Shokat KM, et al. Structure of the human autophagy initiating kinase ULK1 in complex with potent inhibitors. ACS Chem Biol. 2015 Jan 16;10(1):257–261.
- Li H, Kaminski MS, Li Y, et al. Mutations in linker histone genes HIST1H1 B, C, D, and E; OCT2 (POU2F2); IRF8; and ARID1A underlying the pathogenesis of follicular lymphoma. Blood. 2014 Mar 6;123(10):1487–1498.
- Ross CW, Ouillette PD, Saddler CM, et al. Comprehensive analysis of copy number and allele status identifies multiple chromosome defects underlying follicular lymphoma pathogenesis. Clin Cancer Res. 2007 Aug 15;13(16):4777–4785.
- Hu N, Wang F, Sun T, et al. Follicular lymphoma-associated BTK mutations are inactivating resulting in augmented AKT activation. Clin Cancer Res. 2021 Apr 15;27(8):2301–2313.
- Gueldener U, Heinisch J, Koehler GJ, et al. A second set of loxP marker cassettes for Cre-mediated multiple gene knockouts in budding yeast. Nucleic Acids Res. 2002 Mar 15;30(6):e23.
- Longtine MS, Mckenzie A III, Demarini DJ, et al. Additional modules for versatile and economical PCR‐based gene deletion and modification in Saccharomyces cerevisiae. Yeast. 1998;14(10):953–961.
- Feng Y, Ariosa AR, Yang Y, et al. Downregulation of autophagy by Met30-mediated Atg9 ubiquitination. Proc Nat Acad Sci. 2021;118(1):e2005539118.
- Memisoglu G, Eapen VV, Yang Y, et al. PP2C phosphatases promote autophagy by dephosphorylation of the Atg1 complex. Proc Nat Acad Sci. 2019;116(5):1613–1620.
- Wen X, Gatica D, Yin Z, et al. The transcription factor Spt4-Spt5 complex regulates the expression of ATG8 and ATG41. Autophagy. 2020 Jul;16(7):1172–1185.
- Cabrera M, Ungermann C. Chapter thirteen purification and in vitro analysis of yeast vacuoles. Methods Enzymol. 2008;451:177–196.
- Tomashek JJ, Sonnenburg JL, Artimovich JM, et al. Resolution of subunit interactions and cytoplasmic subcomplexes of the yeast vacuolar proton-translocating ATPase (*). J Biol Chem. 1996;271(17):10397–10404.
- Morano KA, Klionsky DJ. Differential effects of compartment deacidification on the targeting of membrane and soluble proteins to the vacuole in yeast. J Cell Sci. 1994;107(10):2813–2824.
- Bartolommei G, Moncelli MR, Tadini-Buoninsegni F, et al. A method to measure hydrolytic activity of adenosinetriphosphatases (ATPases). PLoS One. 2013;8(3):e58615.
- Benjamini YHY. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc. 1995;57(1):289–300.
- Virtanen P, Gommers R, Oliphant TE, et al. SciPy 1.0: fundamental algorithms for scientific computing in Python. Nat Methods. 2020 Mar;17(3):261–272.
- Seabold SPJ. statsmodels: econometric and statistical modeling with python. 9th Python in Science Conference; Austin, TX. . 2010.
- Hunter JD. Matplotlib: a 2D graphics environment. Comput Sci Eng. 2007;9(3):90–95.
- Klionsky DJ, Cueva R, Yaver DS. Aminopeptidase I of Saccharomyces cerevisiae is localized to the vacuole independent of the secretory pathway. J Cell Biol. 1992 Oct;119(2):287–299.
- Huang WP, Scott SV, Kim J, et al. The itinerary of a vesicle component, Aut7p/Cvt5p, terminates in the yeast vacuole via the autophagy/Cvt pathways. J Biol Chem. 2000 Feb 25;275(8):5845–5851.