
ABSTRACT
Macroautophagy/autophagy is a conserved eukaryotic process to mediate the degradation of cell organelles and protein aggregates, which participates in a variety of cellular responses, including immune signal transduction. KDM4D functions as an important histone demethylase to regulate gene transcription by inhibiting histone H3K9 trimethylation. Whether autophagy epigenetically regulates the immune response via modulating the stability and activity of KDM4D remains largely unclear. Recently, we identified TRIM14 (tripartite motif-containing 14) as an epigenetic regulator, which recruits USP14 and BRCC3 to form a regulatory complex, and promotes an inflammation response through inhibiting OPTN-mediated autophagic degradation of KDM4D.
Autophagy is a degradation and recycling system for intracellular materials and components. Depending on whether cargo receptors are required, autophagy can be executed in a bulk degradation process, or selectively degrades specific substrates. Our previous studies revealed that TRIM14 promotes type I interferon signaling as well as non-canonical NFKB/NF-κB pathway through inhibiting cargo receptor SQSTM1-mediated selective autophagic degradation of the DNA sensor CGAS and the transcriptional factor NFKB2, respectively. In this work, we investigated the potential role of TRIM14 in the epigenetic regulation of histone modification, and found that TRIM14 reduces the level of H3K9 trimethylation (H3K9me3) but not other histone modifications, including H3K4me3, histone H2A ubiquitination (H2Aub) or H2Bub [Citation1]. Because TRIM14 is not a histone modifier, we examined the protein levels of several H3K9 demethylases, and found the protein abundance of KDM4D, but not KDM3B, KDM4A or KDM4B, is increased along with ectopic expression of TRIM14. Moreover, TRIM14 does not influence the mRNA level of KDM4D. Consistent with these results, we observed that downregulation of H3K9me3 mediated by TRIM14 is completely abrogated by knocking out of KDM4D.
In studying the detailed mechanism by which TRIM14 stabilizes KDM4D, we found that TRIM14 can directly interact with KDM4D. KDM4D was previously reported to be degraded by the proteasomal pathway. Through a pharmaceutical inhibitor assay, we found that KDM4D can also be degraded through the autophagy pathway and TRIM14 specifically inhibits the autophagic degradation of KDM4D. Overexpression of TRIM14 impairs the interaction between KDM4D and LC3-II, a conjugated form of the autophagosome marker LC3. Cargo receptors are responsible for recognizing specific substrates and delivering them to phagophores for selective degradation. To distinguish which cargo receptor participates in the autophagic degradation of KDM4D, we checked the interaction between KDM4D and several cargo receptors. Although we found that KDM4D can interact with OPTN, SQSTM1 and CALCOCO2, further experiments showed that the protein abondance of KDM4D is increased in OPTN-knockout (KO) cells but not SQSTM1-KO or CALCOCO2-KO cells. Meanwhile, we observed that ectopic TRIM14 can no longer stabilize KDM4D when there is OPTN deficiency. We further demonstrated that TRIM14 also reduces the interaction of OPTN and KDM4D.
It has been revealed that cargo receptors can mainly target substrates by recognizing their ubiquitin signals. We found that the association between OPTN and KDM4D is significantly decreased when the ubiquitination-binding domain (UBD) of OPTN is deleted. Furthermore, overexpression of TRIM14 decreases the ubiquitination of KDM4D, suggesting that TRIM14 might impair the OPTN-KDM4D interaction by inhibiting the ubiquitination of KDM4D. Then, we analyzed different types of poly-ubiquitination of KDM4D and observed that the K63-linked ubiquitination of KDM4D is significantly reduced by ectopic expression of TRIM14 (). We constructed several KDM4D mutants bearing single lysine (K)-to-arginine (R) substitutions at predicated ubiquitination sites in silico, and found that TRIM14 fails to stabilize the KDM4DK472R mutant, whose K63-linked ubiquitination is markedly reduced. We further discovered that TRIM14 recruits two deubiquitinases (DUBs), USP14 and BRCC3, to form a deubiquitination complex, and functions as a bridge to mediate the interaction between KDM4D and USP14-BRCC3, allowing subsequent removal of K63-linked poly-ubiquitin chains on KDM4D. BRCC3 and USP14 might play a redundant role for KDM4D ubiquitination, because KO of either BRCC3 or USP14 does not affect TRIM14-mediated deubiquitination and stabilization of KDM4D. Interestingly, we showed that the fusion protein of the USP domain of USP14 and TRIM14 can stabilize KDM4D alone even in USP14/BRCC3 double-KO cells, showing a new strategy of fusion protein design for regulating selective autophagy.
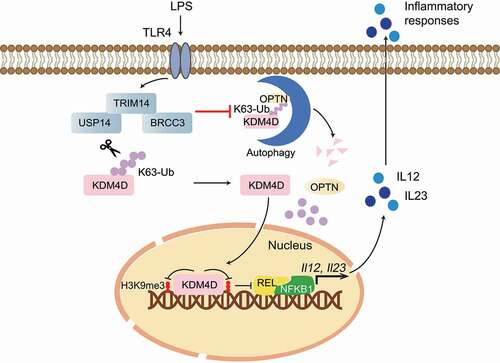
Figure 1. A working model to elucidate that OPTN-mediated selective autophagic degradation of KDM4D epigenetically regulates inflammation. The TRIM14-USP14-BRCC3 complex reduces the K63-linked ubiquitin chains of KDM4D to inhibit OPTN-mediated autophagic degradation of KDM4D, which is responsible for removing the H3K9me3 modification at Il12 and Il23 promoters to promote the expression of proinflammatory cytokines IL12 and IL23 to enhance inflammation. LPS, lipopolysaccharide.
We next investigated how TRIM14 modulates inflammation byusing a mouse experimental autoimmune encephalomyelitis (EAE) model, and found that the deficiency of Trim14 increases H3K9me3 modification at promoters of Il12a, Il12b and Il23a, which limits the expression of these pro-inflammatory cytokines. Compared with wild-type mice, we detected not only less infiltration of CD4+ immune cells, but also less transcription of Il12a, Il12b and IL23a in brain and spinal cord of Trim14−/− mice. Consistently, reduced clinical symptom severity of EAE was observed in Trim14−/− mice, indicating that Trim14 deficiency is beneficial to resolve inflammation and promote resistance to EAE.
In summary, our study suggests that TRIM14 functions as an epigenetic regulator of inflammation. TRIM14 recruits USP14 and BRCC3 to remove the K63-linked ubiquitin chains of KDM4D, therefore preventing KDM4D from interacting with the cargo receptor OPTN for subsequent autophagic degradation. Deficiency of TRIM14 reduces the removal of H3K9me3 at the promoters of Il12 and Il23, thus protecting mice from autoimmune inflammation (). These findings highlight the function of autophagy to regulate immune responses at the epigenetic level and may provide a potential target for inflammation-related diseases.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
Reference
- Liu D, Zhao Z, She Y, et al. TRIM14 inhibits OPTN-mediated autophagic degradation of KDM4D to epigenetically regulate inflammation. Proc Natl Acad Sci U S A. 2022;119(7):e2113454119.