
ABSTRACT
Neurons depend on macroautophagy/autophagy to maintain cellular homeostasis, and loss of autophagy leads to neurodegeneration. To better understand the role of basal autophagy in neurons, we enriched autophagic vesicles from healthy adult mouse brain and performed mass spectrometry to identify cargos cleared by autophagy. We found that synaptic and mitochondrial proteins comprise nearly half of the unique AV cargos identified in brain. Similarly, synaptic and mitochondrial proteins are major cargos for basal autophagy in neurons. Strikingly, we noted a specific enrichment of mitochondrial nucleoids within neuronal autophagosomes, which occurs through a mechanism distinct from damage-associated mitophagy. Here, we discuss the implications of these findings for our understanding of homeostatic mechanisms in neurons and how the age-dependent decline of autophagy in neurons may contribute to the onset or progression of neurodegenerative disease.
Neurons are post-mitotic, long-lived cells with a unique architecture that allows them to form polarized connections, or synapses, with adjoining cells to give or receive information. These properties, in conjunction with high metabolic demands, result in a strong reliance on mechanisms like autophagy to avoid proteotoxicity or the accumulation of damaged organelles. Indeed, neurons in culture under nutrient-replete conditions will begin to die 24 h after autophagy inhibition. Neuronal autophagy is critical and so is tightly regulated in ways distinct from other cell types. In neurons, autophagosomes form constitutively in the distal axon or at synapses. Newly formed autophagosomes then traffic retrogradely; cargo degradation initiates during transport resulting in delivery of the proteolytic products to the soma for recycling. Seminal work has shown that neuron-specific knockout of autophagy is sufficient to induce rapid neurodegeneration, and dysfunctional autophagy is associated with Alzheimer disease, Parkinson disease, and amyotrophic lateral sclerosis. Thus, understanding what cargos are degraded by autophagy may provide clues to the etiology of multiple different neurodegenerative diseases.
In Goldsmith et al. [Citation1], we identified autophagic cargo by employing unbiased proteomics on autophagic vesicles (AVs) isolated from brain tissue by differential ultracentrifugation. AVs include autophagosomes and autolysosomes, with minimal organelle contamination confirmed by immunoblotting, electron microscopy and immunofluorescence. Of note, this approach does not rely on selection using external autophagosome markers that are cleaved during maturation. Half of each AV fraction was incubated with proteinase K to degrade externally associated proteins, such as the motor proteins required for AV trafficking, resulting in the enrichment of AV contents protected within the organelle’s membrane. Comparison of the proteinase K-protected fraction to the total AV fraction provides a snapshot of the proteins that are destined for degradation by autophagy in unperturbed brain samples, as opposed to monitoring the accumulation of proteins following prolonged autophagy inhibition, which is known to be poorly tolerated in neurons and therefore susceptible to alteration by compensatory pathways.
The lack of enrichment of well-characterized autophagy receptor proteins such as SQSTM1/p62, OPTN, NBR1, CALCOCO2/NDP52, and BNIP3L/NIX within AVs was initially surprising. This suggests that under basal conditions, homeostatic autophagy in the brain captures cargo by bulk engulfment of cytoplasm near the forming autophagosome and is not mediated by receptor proteins. Our results are consistent with previous studies in Drosophila that stress-induced pathways such as the PINK1-PRKN/Parkin mitophagy mechanism do not contribute substantially to mitochondrial turnover under basal conditions.
Synaptic proteins have been reported to be autophagy cargo in brain, with this turnover implicated in learning and memory. We found that 18% of the unique cargo identified in brain-derived AVs are synaptic proteins. We validated the presence of the pre-synaptic proteins SYN1 (synapsin I), VAMP2, and SYP (synaptophysin) in both brain-derived and neuron-derived autophagosomes by biochemical fractionation. There was a 3-fold enrichment of SYN1 in AVs compared to total brain input; similarly, SYN1 was a cargo in 60% of axonal autophagosomes based on live imaging in primary hippocampal neurons. Mechanistically, we speculate that autophagy degrades synaptic proteins because of spatial proximity of the forming autophagosome to synaptic sites. Our findings suggest that a key role for autophagy in neurons is to maintain the synapse, but raise further questions. Does this receptor-independent autophagy preferentially remove older synaptic proteins? We show synaptic proteins are AV cargo in neurons, but it is also possible that synaptic vesicles are degraded by glial autophagy in trans; what is the relative contribution of glial to neuronal autophagy? Further, does the engulfment of synaptic proteins change with activity of the neuron? Both pre-synaptic and post-synaptic proteins were equally likely to be cargos within AVs from the brain; how much autophagy is occurring in dendrites relative to axons in vivo? Addressing these questions in the future will provide a more complete picture of how autophagy supports synaptic transmission and neuronal function.
Nearly 20% of the unique cargo identified in autophagic vesicles from brain are mitochondrial proteins, with identifiable mitochondrial fragments found in 50% of the AVs by electron microscopy and fluorescence microscopy. One of the more surprising findings of our work is that mitochondrial DNA (mtDNA)-associated proteins are specifically enriched in basal autophagosomes over other types of mitochondrial proteins. In fact, the nucleoid-associated protein TFAM is more enriched in brain-derived AVs compared to isolated mitochondria from the brain. Live imaging experiments with hippocampal neurons confirmed TFAM is a cargo in 40% of autophagosomes in the axon, whereas the mitochondrial respiratory chain protein COX8A is found in only 20% of autophagosomes. Interestingly, TFAM is enriched in AVs derived from neurons but not from isogenic iPSCs, suggesting this selective degradation of mtDNA by autophagy may be activated during neuronal differentiation.
We hypothesize that mitochondrial fragments containing mtDNA and associated proteins including TFAM become enriched in AVs based on proximity rather than selection (). Because mtDNA replication occurs at mitochondria-ER contact sites, and the ER is the point of initiation for autophagosome formation, the apposition of these two events would increase the likelihood of capture of mtDNA into newly forming autophagosomes. Consistent with this hypothesis, the mitochondrial fission factor DNM1L/Drp1 is required for autophagic engulfment of nucleoids, and MFF (associated with mtDNA replication) but not FIS1 (associated with damage-induced mitophagy) is enriched within the autophagic vesicles. Because our results demonstrate that the turnover of nucleoid-enriched mitochondrial fragments is distinct from receptor-mediated mitophagy of damaged mitochondria, we are investigating how these two processes work together to maintain the mitochondrial network in neurons.
Figure 1. Model of receptor-independent engulfment of cargo in neuronal autophagosomes. We hypothesize that the proximity of the nascent phagophore to synapses and sites of mitochondrial DNA replication and fission leads to increased likelihood of synaptic vesicle and nucleoid-containing mitochondrial fragment capture in the axon of neurons. Within enriched autophagosomes (double membrane pseudo-colored purple) we identify structures corresponding with synaptic vesicles (pseudo-colored yellow) and mitochondrial fragments (pseudo-colored green) by electron microscopy. Live imaging in primary hippocampal neurons suggests that nucleoids marked by TFAM and pre-synaptic proteins such as SYN1 are present in approximately 70% of trafficking autophagosomes in the axon.
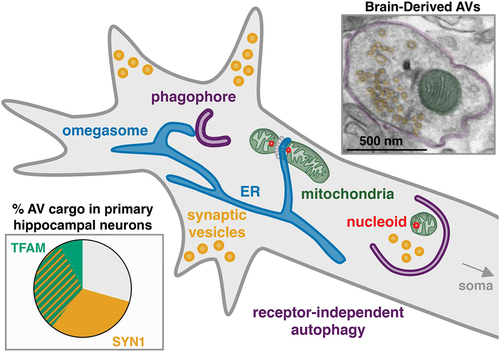
These findings suggest that basal autophagy may contribute to the maintenance of mitochondrial genome copy number. Whether autophagy regulates mtDNA copy number specifically in the axon or in the soma as well, and what impact impaired autophagy has on total neuronal mtDNA copy number remains to be determined. Given the fact that the engulfment of these fragments is not receptor-mediated, it seems unlikely that this process actively removes damaged mtDNA, but it may have an impact on mtDNA mutation rates over long periods of time. Importantly, because neuronal autophagy declines with aging, our results predict an age-dependent accumulation of mtDNA within neurons. As recent research has demonstrated that the release of mtDNA plays a role in neuroinflammation that exacerbates neurodegenerative disease, understanding the role of autophagy in the mitigation of neuroinflammation by regulating the quantity and quality of mtDNA in the neuron is one of our next major research questions.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
Reference
- Goldsmith J, Ordureau A, Harper JW, et al. Brain-derived autophagosome profiling reveals the engulfment of nucleoid-enriched mitochondrial fragments by basal autophagy in neurons. Neuron. 2022;110:1–10. Available from: https://doi.org/https://doi.org/10.1016/j.neuron.2021.12.029