
ABSTRACT
Two autophagy-related (ATG) ubiquitin-like conjugation systems, the ATG12 and ATG8 systems, play important roles in macroautophagy. While multiple duplications and losses of the ATG conjugation system proteins are found in different lineages, the extent to which the underlying systems diversified across eukaryotes is not fully understood. Here, in order to understand the evolution of the ATG conjugation systems, we constructed a transcriptome database consisting of 94 eukaryotic species covering major eukaryotic clades and systematically identified ATG conjugation system components. Both ATG10 and the C-terminal glycine of ATG12 are essential for the canonical ubiquitin-like conjugation of ATG12 and ATG5. However, loss of ATG10 or the C-terminal glycine of ATG12 occurred at least 16 times in a wide range of lineages, suggesting that possible covalent-to-non-covalent transition is not limited to the species that we previously reported such as Alveolata and some yeast species. Some species have only the ATG8 system (with conjugation enzymes) or only ATG8 (without conjugation enzymes). More than 10 species have ATG8 homologs without the conserved C-terminal glycine, and Tetrahymena has an ATG8 homolog with a predicted transmembrane domain, which may be able to anchor to the membrane independent of the ATG conjugation systems. We discuss the possibility that the ancestor of the ATG12 and ATG8 systems is more similar to ATG8. Overall, our study offers a whole picture of the evolution and diversity of the ATG conjugation systems among eukaryotes, and provides evidence that functional diversifications of the systems are more common than previously thought.
Abbreviations: APEAR: ATG8–PE association region; ATG: autophagy-related; LIR: LC3-interacting region; NEDD8: neural precursor cell expressed, developmentally down-regulated gene 8; PE: phosphatidylethanolamine; SAMP: small archaeal modifier protein; SAR: Stramenopiles, Alveolata, and Rhizaria; SMC: structural maintenance of chromosomes; SUMO: small ubiquitin like modifier; TACK: Thaumarchaeota, Aigarchaeota, Crenarchaeota, and Korarchaeota; UBA: ubiquitin like modifier activating enzyme; UFM: ubiquitin fold modifier; URM: ubiquitin related modifier.
Introduction
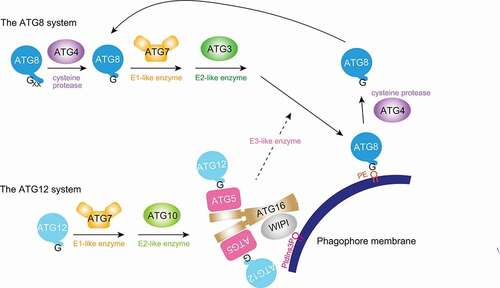
Macroautophagy, hereafter referred to as autophagy, is conserved across eukaryotes. The ATG12 and ATG8 systems are two ubiquitin-like conjugation systems that play important roles in autophagosome formation and maturation. The ATG12 system utilizes the E1-like enzyme ATG7 and the E2-like enzyme ATG10 to conjugate the ubiquitin-like protein, ATG12, to its substrate, ATG5 (ATG“X” [all in uppercase letters] is used to generally refer to ATG“X” homologs in any organisms including yeasts, while the species-specific nomenclature is used when referring to a particular species, such as Atg“X” in yeast). Together with the ATG16 dimer, it forms the ATG12–ATG5-ATG16 complex. The ATG8 system uses the same E1-like enzyme and a different yet evolutionarily related E2-like enzyme, ATG3. Finally, the ATG12–ATG5-ATG16 complex functions as an E3-like enzyme that facilitates ATG8 conjugation to phosphatidylethanolamine (PE) in the autophagic membranes [Citation1]. ATG8 needs to be processed by the cysteine protease ATG4 first to expose its C-terminal glycine for conjugation [Citation2], while there is no such need for ATG12 as it ends in glycine (). ATG12 is conjugated to a central lysine residue in ATG5, whereas ATG8 is conjugated to an amide group in PE.
Figure 1. During autophagy, two ubiquitin-like ATG conjugation systems function together to conjugate ATG8 to the phagophore membrane. The ATG12 system conjugates ATG12 to ATG5, and the ATG12–ATG5-ATG16 complex functions as an E3-like enzyme facilitating the conjugation of ATG8 to PE. Conjugation requires the C-terminal glycines in ATG8 and ATG12. While ATG12 ends in glycine, typical ATG8 needs C-terminal processing by the cysteine protease ATG4 to expose it. ATG4 also deconjugates ATG8 from the membrane for recycling purposes. ATG3 and ATG10, and ATG8 and ATG12 are evolutionarily related. WIPI (or Atg21) family proteins recruit ATG16 to autophagic membranes. PE, phosphatidylethanolamine. PtdIns3P, phosphatidylinositol-3-phosphate. WIPI, WD repeat domain, phosphoinositide interacting.
It is thought that the eukaryotic ubiquitin and ubiquitin-like proteins, including URM1 (ubiquitin related modifier 1), UFM1 (ubiquitin fold modifier 1), NEDD8 (neural precursor cell expressed, developmentally down-regulated gene 8), SUMOs (small ubiquitin like modifier), ATG8 and ATG12, form a monophyletic group that evolved from prokaryotic sulfur transfer proteins such as ThiS and MoaD [Citation3–5]. URM1 in eukaryotes and SAMP1/SAMP2 (small archaeal modifier protein) in archaea are dual-function proteins capable of both protein conjugation and sulfur transfer [Citation6–12] and are at an evolutionary position close to the ancestral protein [Citation13,Citation14]. ATG8 and ATG12 have limited sequence homology to ubiquitin, and form their own cluster on the phylogenetic trees [Citation4,Citation15–17]. However, it is not clear which is more ancestral, ATG8 or ATG12. More recently, the hypothesis that eukaryotes emerged from within an archaeal group known as Asgard has gained strong support [Citation18–20], and complete ubiquitin-like systems (with E1-, E2-, and E3-like enzymes) evolutionarily close to those in eukaryotes have been identified in Aigarchaeota and Asgard archaea [Citation18, Citation19, Citation21–23]. Including homologs from these archaeal groups will help provide a better evolutionary context to the emergence of the ATG conjugation systems during evolution.
Within eukaryotes, the ATG conjugation systems are most well-understood in metazoan and yeast species, followed by plants, Dictyostelium and pathogenic protists such as Plasmodium, Toxoplasma, and Trypanosoma [Citation1,Citation24–27]. On the one hand, ATG8 and ATG4 show extensive duplications and functional diversifications in Metazoa, Dictyostelium, plants, Alveolata, and Discoba [Citation28–33]. On the other hand, ATG10 and ATG12 appear to be less conserved in Stramenopiles, Alveolata and Rhizaria (SAR) and fungi [Citation34]. In Plasmodium, Toxoplasma and yeast species such as Komagataella, the E2-like enzyme ATG10 and the C-terminal glycine of ATG12 that are required for the covalent conjugation were lost during evolution. Instead, non-covalent interactions are in place to ensure normal function to promote ATG8–PE conjugation. However, there is vast diversity within eukaryotes [Citation35,Citation36], and many branches remain unexplored. Whether such non-covalent system or other non-canonical systems exist beyond the aforementioned lineages, is unknown.
In order to understand the evolutionary diversity of the ATG conjugation systems in eukaryotes, we constructed a transcriptome database consisting of 94 species spanning most of the major eukaryotic groups [Citation36]. As many of these species lack high-quality reference genomes, de novo transcriptome assembly provides a powerful means to study the coding genes [Citation37]. Systematic identification and annotation of the ATG conjugation system components uncovered diversifications in both the ATG12 and ATG8 systems. We divided the ATG conjugation systems into six major types and discuss their implications. Our study offers a broad view of the evolution and diversity of the ATG conjugation systems in eukaryotes.
Results and discussion
Construction of the transcriptome database and identification of the ATG conjugation system components
In order to properly sample the diversity within eukaryotes, we manually selected 94 species covering most of the major clades in eukaryotes, including TSAR (Telonemia, Stramenopiles, Alveolata and Rhizaria), Haptista (Centrohelida and Haptophyta), Cryptista, Archaeplastida (Glaucophyta, Chlorophyta and Rhodophyta), Ancoracysta, Amorphea (Breviates, fungi, Holozoa and Amoebozoa), CRuMs (collodictyonids, rigifilids, and Mantamonas), Discoba, Metamonada, Malawimonadida, Ancyromonadida and Hemimastigophora [Citation36,Citation38–47] (Fig. S1). The exact phylogenetic placement of some clades is an area of active research, and we used the consensus eukaryotic tree from Burki et al. 2020 [Citation36] in this study. As high-quality reference genomes and gene annotations may not exist for many of these organisms, we assembled the transcriptome de novo from RNA-sequencing data (source data listed in Table S1; seven and eight species had proteome and pre-assembled transcriptome data downloaded directly, respectively) using Trinity [Citation48]. Subsequent coding region prediction and translation were conducted by TransDecoder [Citation49]. The mean and median BUSCO scores [Citation50], a measure of the completeness of the assembled transcriptomes, are 75.9 and 79.5, respectively. We supplemented this custom database with sequences from NCBI protein and UniProt [Citation51,Citation52] if a species is available in these databases to make them as complete as possible.
Overall, de novo transcriptome assembly was very useful in studying the gene contents of non-model organisms. However, some species (e.g., Dysnectes, Rhynchopus) in our study had low BUSCO scores and few sequences deposited in the NCBI protein and UniProt databases. Therefore, some sequences may still be missing from our analyses for these species.
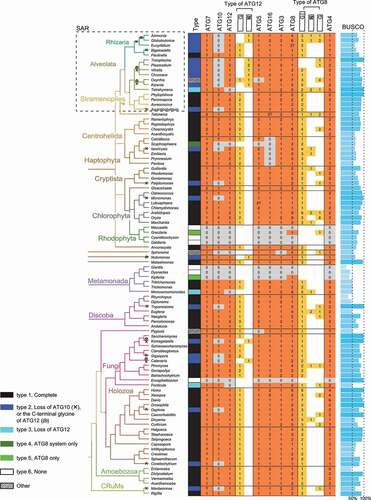
The pipeline to systematically identify and annotate ATG conjugation system components in these species is shown in Fig. S2. There are two main challenges associated with the analysis. First, because ATG12 and ATG10 are evolutionarily related to ATG8 and ATG3, respectively [Citation4,Citation5], the pipeline needs to be able to differentiate these sequences. This was achieved by considering conserved domains, reciprocal search results, and phylogenetic clusters if the former two failed (see Methods for detail). Second, because our dataset consists of a diverse selection of species, the same query sequence, e.g., from the budding yeast Saccharomyces cerevisiae, may fail to identify all homologs. Therefore, two rounds of analysis were conducted, where the second-round analysis used homologs identified from the first round as the new query sequences (see Methods for detail). In order to increase the sensitivity of the homology search, both DELTA-BLAST and HMMER [Citation53,Citation54] were used for ATG10 and ATG12 sequences. Because ATG16 contains a coiled-coil region and a WD40 domain, both of which were difficult for homology search, additional search rounds were carried out. Sequence clustering by Graph Splitting [Citation55] was able to distinguish ATG16 homologs (Fig. S3, green circles) from non-homologs (Fig. S3, white and gray circles). Five putative homolog sequences were removed (Fig. S3, white circles within the area surrounded by the green dashed line) because better sequences existed in those species. All candidate sequences were subject to manual inspection and quality control. Our analysis identified a total of 94 ATG7, 69 ATG10, 94 ATG12, 98 ATG5, 97 ATG16, 107 ATG3, 223 ATG8, and 155 ATG4 homologs in the 94 eukaryotic species (). The alignment of these sequences (columns with many gaps skipped) is in Data S1.
Figure 2. The distribution of ATG7, ATG10, ATG12, ATG5, ATG16, ATG3, ATG8, and ATG4 homologs in 94 eukaryotic species. For simplicity, only the genus name of the species is shown, and the full name can be found in Table S1. Independent ATG10 loss or the loss of ATG12ʹs C-terminal glycine, which occurred at least 16 times in 21 species, are labeled by a cross or a swirl, respectively, on the phylogenetic tree of the species to the left. The first column indicates the type of the ATG conjugation system, with the legend at the bottom left (see ). Columns in Orange indicate the number of homologs identified (the number of homologous sequences at 80% similarity threshold). “?” means that the sequence is incomplete, i.e., either short or missing key residues including catalytic residues in ATG7, ATG10, ATG3 or ATG4, and the lysine to which ATG12 is conjugated in ATG5. Columns in yellow indicate ATG12 and ATG8 homologs further classified by their C termini into canonical (conserved glycine followed by additional residues), without C-terminal glycine, or no extension beyond glycine. BUSCO scores, which is a measure of the completeness of the custom transcriptome dataset ranging from 0% to 100%, are displayed to the right.
The distribution of the ATG conjugation system components in eukaryotes
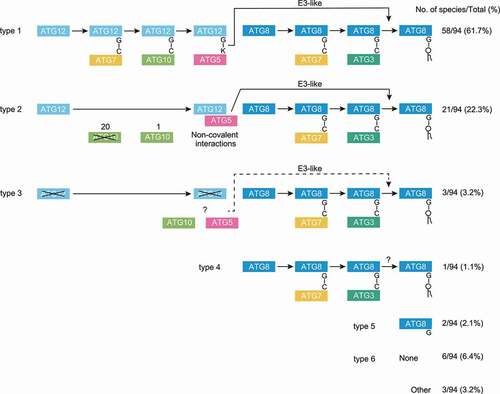
The number of ATG7, ATG10, ATG12, ATG5, ATG16, ATG3, ATG8, and ATG4 homologs among the 94 eukaryotic species, together with the BUSCO scores of the assembled transcriptomes, are shown in . The C-terminal status of ATG12 and ATG8 homologs (with or without the conserved glycine and any following additional sequence), as determined by multiple sequence alignment or Hidden Markov Model-based alignment, are also shown. Our survey demonstrates that the ATGs related to the conjugation systems are conserved in all eukaryotic clades. Although these ATGs are not conserved in Microsporidia, Rhodophyta and some species in Metamonada, the phylogenetic tree suggests that they have lost ATGs secondarily (discussed below). The data strongly indicates that the ATG conjugation systems have already been acquired in the last eukaryotic common ancestor. Overall, the 94 eukaryotic species can be classified into six major types by the completeness of the ATG12 and ATG8 systems: (type 1) complete; (type 2) no ATG10 or no C-terminal glycine in ATG12 (non-covalent ATG12 system); (type 3) intact ATG10 and ATG5 but no ATG12; (type 4) ATG8 system only (with ATG3); (type 5) ATG8 homologs only (without ATG3); (type 6) none (). Three species, Oxyrrhis (Alveolata), Spironema (Hemimastigophora), and Pygsuia (Breviates), could not be categorized into these six types and are instead noted as “Other”.
Figure 3. The ATG conjugation systems in the 94 eukaryotic species can be classified into six major types, with a few exceptions. The six types are: (type 1) Complete; (type 2) Loss of either ATG10 or the C-terminal glycine of ATG12, resulting in possible non-covalent interactions between ATG12 and ATG5; (type 3) Loss of ATG12 but not ATG10 or ATG5; (type 4) ATG8 system only; (type 5) ATG8 homolog only; and (type 6) none. The number of species out of the total 94 species is shown for each group, and the type number is labeled on the left. Species in type 6 lacks not only the ATG conjugation system components but also most of other ATG proteins, and presumably lacks the autophagy pathway altogether. For the purpose of classification, “?” was treated as normal.
The conservation of LC3-interacting region (LIR) motifs in ATG12 (3D-LIR) [Citation56], Atg3 (WEDL in S. cerevisiae) [Citation57], and ATG4 (C-terminal LIR [cLIR] and ATG8–PE association region [APEAR]) [Citation58], is displayed in Fig. S4. The 3D-LIR motif in ATG12 may be partially conserved in a wide range of species, while the WEDL motif in Atg3 is limited to the budding yeast and closely-related species as it resides in a loop mostly specific to fungi. The LIR motifs in ATG4 are also mostly conserved and will be discussed later.
The frequent transition from the covalent to non-covalent ATG12 system
Independent loss of ATG10, the E2-like enzyme for the ATG12 system, occurred at least 15 times independently (in 20 species) among the species we surveyed (, type 2). In addition to the previously reported events within SAR and fungi [Citation34], ATG10 loss also occurred sporadically in a wide range of lineages (). Because ATG10 is necessary for the covalent conjugation between ATG12 and ATG5, non-covalent interactions may be more widespread than previously thought. We were also able to refine the timing of ATG10 loss in the SAR group. Most Alveolata and Rhizaria species, but not Stramenopiles, lost ATG10, even though Alveolata and Stramenopiles are more closely related to each other, suggesting that the loss of ATG10 was not an ancestral feature of SAR.
Another feature that often accompanies ATG10 loss in species that switched to the non-covalent ATG12 system is the loss of the C-terminal glycine in ATG12, which is a key residue required for conjugation [Citation34]. In most species, ATG10 loss appears to be an early event. However, there is an exception: Nutomonas (Ancyromonadida) has intact ATG10 but ATG12 lacking the C-terminal glycine. Emiliania’s ATG12 contains possible GC-AG splice sites, non-canonical splice sites suggested to exist in this organism [Citation59], near its C terminus , resulting in canonical ATG12 if spliced and non-canonical ATG12 if retained, but the transcriptomic evidence for the canonical isoform is stronger, so we counted it as canonical. Therefore, it is possible that the order of events leading up to non-covalent interactions could be flexible, but the loss of ATG10 occurred earlier in most cases. This could be because gene-deactivating mutations that ultimately lead to gene loss through pseudogenization [Citation60], such as nonsense, frameshift or splice site mutations, can happen anywhere in the gene. Therefore, in a scenario where selection pressure to maintain covalent conjugation is relaxed, the chance of random mutations leading to ATG10 loss is higher than the loss of the C-terminal glycine in ATG12, which requires a point mutation.
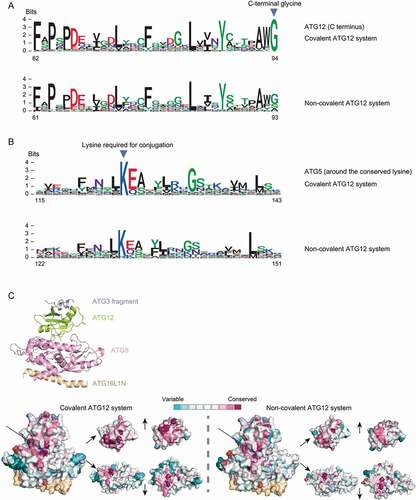
In order to understand if the development of non-covalent interactions between ATG12 and ATG5 requires specific sequence features, we divided the ATG12 and ATG5 homologs identified in our study into the covalent and non-covalent groups and plotted the sequence logos of each group ( for ATG12 and ATG5, respectively). The sequence logos are very similar in general (sequences other than the region shown in are also very similar [data not shown]), suggesting that no specific motifs are required for non-covalent interaction. In agreement with this observation, conservation scores for ATG12 and ATG5 mapped onto the human structure (PDB: 4naw) also show no significant difference between the covalent and the non-covalent systems (other than the C-terminal glycine) ()). It should be noted that the lysine residue in ATG5, which is the conjugation site in the covalent ATG12 system, is still conserved in the non-covalent system with two exceptions (Toxoplasma and Karenia) in which lysine has been replaced with arginine ()). Thus, the positively charged amino acids (lysine is preferred) may contribute to the non-covalent interaction. Previously, we found that the same positions that are important for the non-covalent interactions between ATG12 and ATG5 in Toxoplasma (a species with the non-covalent ATG12 system) also harbor weak non-covalent interactions in human (a species with the covalent ATG12 system) [Citation34]. Taken together, we speculate that mutations that are either subtle or not very well-conserved led to strengthening the existing interfaces or the creation of new interfaces, which eventually developed into the non-covalentsystem. In summary, loss of ATG10 or the C-terminal glycine of ATG12 represents the most common alternative ATG conjugation system type in this study.
Figure 4. Comparison of amino acid sequences of ATG12 and ATG5 homologs between covalent and non-covalent ATG12 systems. Sequence logos of the C termini of ATG12 homologs (A) and the region around the lysine (conjugated to ATG12 in the covalent system) in ATG5 homologs (B) are displayed, which were collected from the covalent and possibly non-covalent ATG12 systems (type 1 and type 2 in ), where the non-covalent system is defined by the lack of ATG10 homolog or the lack of the C-terminal glycine in ATG12. Positions corresponding to the C-terminal glycine in ATG12 and the conserved lysine in ATG5 are shown by arrowheads. Numbers at the bottom are positions in the multiple sequence alignment (columns with more than 20% gap are removed). (C) Conservation scores calculated by ConSurf mapped onto the human ATG12–ATG5-ATG16L1 complex together with the flexible region of ATG3 (PDB: 4naw). A cartoon model showing each protein is at the top, and the surface models color-coded by conservation levels are at the bottom. A front view of the structure is in the bottom left, and the interacting interfaces of ATG12 and ATG5 are in the bottom right. The C-terminal glycine of ATG12 and the lysine required for conjugation in ATG5 are labeled with arrows and dashed circles. Two possible ATG12 sequence fragments in the covalent ATG12 system that have incomplete C termini, and four possible ATG5 sequence fragments in the covalent ATG12 system that lack the conserved central region (“?” in ) were not included in this plot.
Loss of the ATG conjugation components
Tetrahymena (Alveolata), Monocercomonoides (Metamonada), and Fonticula (fungi) have ATG10 but no ATG12 homologs (, type 3). On the other hand, Tetrahymena contains five ATG8 homologs with exact functions unknown. Because ATG12 and ATG8 are evolutionarily related, it is conceivable that some of these ATG8 homologs might act as functional substitutes for ATG12 and are catalyzed by ATG10. This idea is consistent with instances of ATG8ylation (ATG8 conjugation to proteins) discovered recently [Citation61–63]. The same possibility exists for the other two species, Monocercomonoides and Fonticula, as they also contain multiple ATG8 homologs each, but as their BUSCO scores are low, the lack of ATG12 may also be due to incomplete transcriptome assembly.
Scyphosphaera (Haptophyta) only had the ATG8 system (, type 4). Normally, the ATG12–ATG5 complex functions as an E3-like enzyme for ATG8–PE conjugation both by recruiting ATG3 to the phagophore membrane and by allosterically activating the E2-like enzyme ATG3 [Citation64–67]. ATG8–PE conjugation might be able to proceed without an E3-like enzyme in this species (discussed later).
Gracilaria (Rhodophyta) and Kipferlia (Metamonada, close to Giardia and Dysnectes) have an ATG8 homolog despite a complete lack of other ATG conjugation system proteins (, type 5). Some ATGs outside the ATG conjugation systems can be found for these species, including possible ATG1 homologs for Gracilaria, and possible ATG1, ATG9, VPS15, VPS34 and ATG18 homologs for Kipferlia. Previous research also suggested that species in Rhodophyta and Giardia have TOR (target of rapamycin) [Citation30,Citation32]. Threrefore, despite partial genome reduction, these species, especially Kipferlia, retained some ATG proteins that may work with unlipidated ATG8 in non-autophagy pathways [Citation68–73]. However, in Gracilaria ATG8, the C-terminal glycine, which is a key residue for conjugation, is conserved. It is therefore also possible that this homolog is catalyzed by enzymes from other ubiquitin-like conjugation systems. The Kipferlia sequence has an incomplete C terminus, and the N-terminal fragment is identical to some plant proteins.
Consistent with previous studies [Citation30,Citation32,Citation33], we found no ATG homologs in Encephalitozoon (fungi), Metamonads including Giardia and Dysnectes, and most species in Rhodophyta (, type 6). The loss of ATG genes, and presumably the autophagy pathway, is likely a result of the extensive genome reductions reported in these species [Citation74–76], possibly related to adaptations to either a parasitic lifestyle or extreme environments.
ATG8 homologs ending in glycine
Next, we examined the amino acid sequence of the C-terminal region of ATG8. All species with ATG8 homologs have at least one canonical ATG8 protein (i.e., with a few amino acids extending beyond the C-terminal glycine), except for Toxoplasma, Plasmodium, and Gracilaria that only have ATG8 homologs ending in glycine (). Because ATG4 is the protease responsible for cleaving residues beyond glycine, we checked if ATG4 is conserved in these species, focusing on the catalytic residues and the ATG8-interacting regions. While the catalytic residues are conserved in Toxoplasma and Plasmodium ATG4, two ATG8-interacting LIR motifs [Citation58,Citation77,Citation78] that are well-conserved among the eukaryotic species are absent. However, Toxoplasma and Plasmodium ATG4s contain multiple putative (i.e., predicted) LIR motifs at other locations. Given the above results, it can be speculated that perhaps Toxoplasma and Plasmodium’s ATG4s have only the deconjugation/delipidation activity but not the C-terminal processing activity. However, Toxoplasma ATG4 has in fact been shown to be capable of both activities [Citation79]. Therefore, ATG8 ending in glycine may have developed because the decoupling of ATG8 and ATG4 at the C-terminal processing step offers some unique advantage for this species. Along with the canonical ATG8 protein, 20 eukaryotic species have additional ATG8 homologs ending in the conserved glycine like Toxoplasma and Plasmodium ATG8. These ATG8 homologs might also be recognized by ATG4s only at the delipidation step and need to have exposed glycine for conjugation.
There are instances where a species have multiple ATG4s but only one ATG8 or vice versa (). In general, the relationship between ATG8 and ATG4 can be complicated, for example different ATG4s can have different C-terminal processing and delipidation activities, and even ATG8-independent functions as recently proposed by Nguyen et al. 2021 [Citation62].
Atypical ATG8 homologs without the C-terminal glycine or with a C-terminal transmembrane domain
We found that more than 10 species have ATG8 homologs without the conserved C-terminal glycine. All of them possess canonical ATG8 homologs with the C-terminal glycine and additional sequences. Therefore, the ATG8 homologs without the C-terminal glycine would have non-canonical conjugation-independent functions. In fact, it is well established that even canonical Atg8 has conjugation-independent functions such as regulating vacuole morphology and function in yeasts, and larval midgut elimination during development in Drosophila [Citation68–73]. Thus, some species might have evolved ATG8 homologs specialized for these conjugation-independent processes.
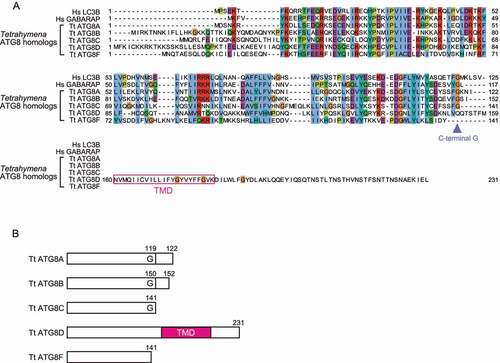
Normally, ATG8 is not a transmembrane protein and is only anchored to the membranes through conjugation to membrane phospholipids with the help of the ATG conjugation systems. However, of the five ATG8 homologs identified in Tetrahymena (Alveolata; ciliate), one contains a predicted transmembrane domain at the C terminus (ATG8D: TTHERM_00526360; multiple sequence alignment and schematic models in ), respectively). As there is no glycine before the predicted transmembrane domain, this domain cannot be cleaved by ATG4. Therefore, ATG8D may be constitutively associated with the membrane independent of other ATG proteins. This could be an adaptation that simplifies the conjugation process if no reversible spatiotemporal regulation is necessary. We also identified an ATG8 homolog with a transmembrane domain in another ciliate Pseudocohnilembus (PPERSA_04697), indicating that this novel type of ATG8 homolog should be conserved in some ciliates and functional. Of the remaining four ATG8 homologs in Tetrahymena, ATG8A (TTHERM_00522490), ATG8B (TTHERM_00037460), and ATG8C (TTHERM_000780499) possess the conserved C-terminal glycine, and ATG8F (TTHERM_00500940) lacks the C-terminal glycine. Both ATG8A and ATG8B are important for programmed nuclear degradation in Tetrahymena, while only ATG8A is required for survival during starvation [Citation80].
Figure 5. Five ATG8 homologs in the ciliate Tetrahymena thermophila. (A) One of the TtATG8 sequences, TtATG8D, has a predicted transmembrane domain (the pink rectangle). The position of the conserved C-terminal glycine is shown by the arrowhead. Multiple sequence alignment is colored using the Clustal X color scheme (blue for hydrophobic residues, red for positively charged residues, purple for negatively charged residues, green for polar residues, pink for C, Orange for G, yellow for P, cyan for aromatic residues, and white for non-conserved positions). (B) Schematic models showing the C-terminal status of the TtATG8 homologs. TMD, transmembrane domain; G, the C-terminal glycine; Hs, Homo sapiens; Tt, Tetrahymena thermophila.
The conservation of ATG16 homologs in eukaryotes
ATG16 homologs could not be found in the Haptophyta species, including Scyphosphaera (type 4) and Emiliania. As part of the ATG12–ATG5-ATG16 complex, ATG16 is required for the correct localization of the ATG12–ATG5 conjugate to the site of autophagosome formation in yeast and mammalian cells [Citation81,Citation82], via both interactions with the Atg1/ULK complex and with membrane directly [Citation83–85]. Despite a lack of ATG16, autophagy-like process has been reported for Emiliania [Citation86], suggesting that other ATGs may have developed ways to compensate for ATG16ʹs function. Human ATG16L1 and ATG16L2 contain WD40 domains at the C termini while Saccharomyces Atg16 does not. Most ATG16 homologs we identified also contain a WD40 domain except for Saccharomyces, Komagataella, Schizosaccharomyces, and maybe Plasmodium, Acanthocystis and Mantamonas. Therefore, even though the WD40 domain is not required for autophagy, ATG16 with the WD40 domain is the ancestral form.
The evolution of E2-like enzymes ATG10 and ATG3, and the ubiquitin-like proteins ATG12 and ATG8
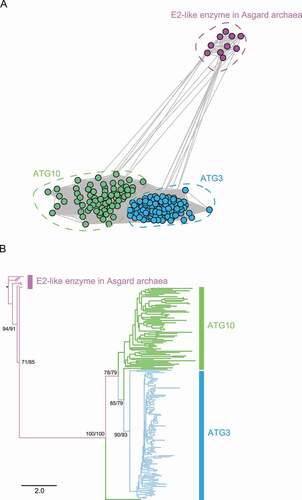
The ATG12 and ATG8 systems share a common ancestor with the ubiquitin and ubiquitin-like conjugation systems, but the evolutionary process behind their emergence and divergence is not well-understood. In order to understand the evolution of the ATG conjugation systems, we reconstructed the phylogenetic trees for the E2-like enzymes ATG10 and ATG3, and the ubiquitin-like proteins ATG12 and ATG8 identified in this study. As our dataset covered most eukaryotic clades, we used homologs from Asgard, an archaeal group from which eukaryotes are hypothesized to have emerged [Citation18,Citation20], as the outgroup. Phylogenetic analysis was conducted using both Graph Splitting [Citation55] ()) and IQ-TREE [Citation87] ()). Graph Splitting is good at identifying and displaying evolutionary relationships as interconnected clusters and has superior performance with remote homologs in large superfamilies, and IQ-TREE is a commonly used maximum likelihood-based method. In the Graph Splitting clustering network ()), each circle represents a sequence, and each line represents a pairwise alignment with its length inversely related to the similarity. Both methods divided ATG3 and ATG10 sequences into two separate clusters, both of which are distant from the Asgard E2-like enzymes, queried using human UBE2M (ubiquitin conjugating enzyme E2 M) (), suggesting that ATG3 and ATG10 already split in the eukaryotic ancestor, which is also consistent with the loss of ATG10 being secondary.
Figure 6. Phylogenetic analysis of the E2-like enzymes ATG10 and ATG3, with UBE2M homologs in the Asgard archaea as the outgroup. The results of Graph Splitting (A) and IQ-TREE (B), which are phylogenetic reconstruction methods relying on spectral clustering and maximum likelihood, respectively, are shown. In the Graph Splitting clustering network (A), each circle represents a sequence and each line represents an alignment between two sequences. The (unrooted) maximum likelihood phylogenetic tree (B) is rooted manually using the Asgard sequences, and statistical support (SH-aLRT/ultrafast bootstrap) is shown for the major branches.
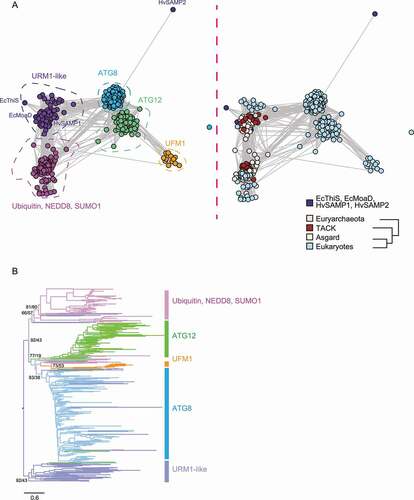
ATG8 and ATG12 belong to the ubiquitin-like family. This family is believed to have originated from sulfur transfer proteins in prokaryotes such as ThiS and MoaD, and members span all three domains of life [Citation3,Citation4]. To clarify the evolutionary relationships between ATG8 and ATG12 and other members in this family, we included ThiS and MoaD from E. coli, all ATG8 and ATG12 homologs identified in this study as well as archaeal (Asgard, Thaumarchaeota, Aigarchaeota, Crenarchaeota and Korarchaeota [TACK] and Euryarchaeota) and eukaryotic URM1, ubiquitin, NEDD8, SUMO1 and UFM1 homologs in the analysis. Graph Splitting divided these proteins into four groups, the URM1-like group including SAMP1, MoaD, and ThiS, the ubiquitin-like group including ubiquitin, NEDD8, and SUMO1, the ATG group including ATG8 and ATG12, and the UFM1 group ()). The UFM1 group appears to be close to ATG12, but its position in the phylogenetic tree is not very stable and thus difficult to define ()) (even though the bootstrap value of the UFM1 branch is not nessasarily lower than other branches, the UFM1 group has a tendency to localize to different positions in different analysis runs). The ubiquitin-like group already separated from the likely ancestral URM1-like group in prokaryotes [Citation12,Citation23]. Since there are not many connections between the ATG group and the ubiquitin-like group, it is possible that they evolved from the ancestral protein separately. The maximum likelihood phylogenetic tree ()) also showed that, apart from a few sequences that ended up in between, the ubiquitin-like group and the ATG group shared a short branch but mostly evolved in parallel.
Figure 7. Phylogenetic analysis of the ubiquitin-like family proteins. (A) Graph Splitting divided the ubiquitin-like family into four groups, the URM1-like group including Escherichia coli ThiS, MoaD, and Haloferax volcanii SAMP, the ubiquitin-like group including ubiquitin, NEDD8 and SUMO1, the autophagy-like group including ATG8 and ATG12, and the UFM1 group. The ubiquitin-like and the autophagy-related group already separated in prokaryotes. The circles, representing sequences, are colored by gene group (left) and by taxonomy (right). (B) The maximum likelihood phylogenetic tree by IQ-TREE shows that the ubiquitin-like and the autophagy-related group shared only a very short branch and mostly evolved in parallel. A total of 163 URM1, ubiquitin, NEDD8, and SUMO1 homologs and 315 ATG8 and ATG12 homologs are included in the plot. The (unrooted) phylogenetic tree is manually rooted by the URM1-like sequences, and statistical support (SH-aLRT/ultrafast bootstrap) is shown for the major branches. Ec, Escherichia coli; Hv, Haloferax volcanii. TACK, Thaumarchaeota, Aigarchaeota, Crenarchaeota, and Korarchaeota.
We also reconstructed the phylogenetic tree for the E1-like enzymes (Graph Splitting network in Fig. S5A and maximum likelihood phylogenetic tree in Fig. S5B). Consistent with the finding in the phylogenetic tree of the ubiquitin-like proteins, more connections exist between ATG7 and MOCS3 (molybdenum cofactor synthesis 3; E1-like enzyme for URM1) than between ATG7 and UBA1–3 (ubiquitin like modifier activating enzyme 1–3; E1-like enzymes for ubiquitin, SUMO1, and NEDD8) in the Graph Splitting network, and the maximum likelihood tree also showed that the ATG7 group and the UBA1–3 group mostly evolved independently. These results further support the hypothesis that the ATG conjugation systems evolved separately from the ubiquitin-like systems. Previous evolutionary and phylogenetic studies also suggest that ATGs are unlikely to have evolved from eukaryotic ubiquitin and ubiquitin-like proteins such as NEDD8 and SUMO, as they are on different branches that separated early on [Citation4,Citation16]. Three studies of the E1-like enzymes that include a wide range of bacterial and archaeal homologs as well as ATG7 also support this notion in general, although they disagree on the exact position of ATG7 in the phylogenetic trees [Citation19,Citation22,Citation88].
Finally, we examined which is more ancestral, the ATG12 or ATG8 systems. In the Graph Splitting network ()), the ATG8 group appears to be closer to the ancestral URM1-like group, with more connections in-between than the ATG12 group. Furthermore, there is no species with ATG12 but without ATG8 among the 94 eukaryotic species we examined in this study (). Therefore, it is possible that the ATG8 system was more ancestral, and the ATG12 system formed by duplications. This possibility is consistent with ATG8 being the functionally more important, downstream system. If true, such an evolutionary scenario suggests that the ancestral ATG8-like system may be able to function without the E3-like enzymes. In fact, the present-day species such as yeast, Toxoplasma, and Arabidopsis can form lipidated ATG8 without the ATG12–ATG5-ATG16 complex in vitro, albeit at a lower efficiency [Citation34,Citation64,Citation89].
Conclusion
While autophagy is thought to be generally conserved in eukaryotes, the underlying systems are subject to modifications and refinements in different clades. In this study, we focused on the ATG conjugation systems. Most of the 94 species we examined have the ATG conjugation systems conserved, indicating that these systems were already present in the last eukaryotic common ancestor. We established six major types of classification for the ATG conjugation systems, ranging from complete to none. The most common type besides the full set is the possible non-covalent ATG12 system, which is more widely distributed than previously found . We also found species with only the ATG8 system, which might be functional without the ATG12 system. Furthermore, there are atypical ATG8 homologs that are unlikely to be conjugated to phospholipids or other molecules, suggesting the functional diversification beyond lipid conjugation. Our phylogenetic analyses suggest that the ATG12 and ATG8 systems already separated in the eukaryotic ancestor. Overall, we reveal the spectrum of diversity in the ATG conjugation systems within eukaryotes and suggest possible evolutionary paths for the emergence of these systems.
Materials and methods
Selection of species and the species phylogenetic tree
We selected 94 species from 21 clades in the eukaryotic tree [Citation36]. The list of species and link to the source RNA-sequencing data for the de novo transcriptome assembly (or proteome data for seven species and pre-assembled transcriptomes for eight species) are in Table S1. Among the 94 species, proteome data was downloaded directly for Plasmodium, Toxoplasma, Entamoeba, Trypanosoma, Trichomonas, Giardia, and human, and pre-assembled transcriptome data was downloaded for Chromera, Karenia, Oxyrrhis, Vitrella, Paulinella, Danio, Drosophila, and Dictyostelium. We assembled the transcriptome de novo in this study for the remaining 79 species. To make the species phylogenetic tree displayed on the left side of , the evolutionary relationships among clades were taken from Burki et al. 2020 [Citation36] and the within-clade phylogenies were taken from the following papers: Alveolata [Citation90], Amoebozoa [Citation91], Centrohelida, Cryptista and Haptophyta [Citation41], Chlorophyta [Citation92,Citation93], Discoba [Citation94], fungi [Citation95–97], Holozoa [Citation95,Citation98,Citation99], Metamonada [Citation100], Rhizaria [Citation101,Citation102], Stramenopiles [Citation103] and Rhodophyta [Citation104].
De novo transcriptome assembly
After RNA-sequencing data was downloaded from either the Sequence Read Archive [Citation105] or the Marine Microbial Eukaryote Transcriptome Sequencing Project [Citation106], reads were subjected to quality filtering by fastp [Citation107] v0.19.4 with options “-5 5 − 3 5 -M 20”. Then, the transcriptome was assembled de novo using Trinity v2.8.4 [Citation48] with the options “--seqType fq --max_memory 100G --left FILE_1.fastq --right FILE_2.fastq --CPU 20” and translated with TransDecoder v5.5.0 [Citation49] with options “-m 50 -G universal” (-G for genetic code was set to “Ciliate” for Tetrahymena). There are two steps in TransDecoder, namely “LongOrfs” which translates all possible reading frames and “Predict” which applies quality control and identifies the most likely reading frame. We used the output file from “LongOrfs” as the custom database, but manually examined any candidate sequence not predicted as the most likely reading frame, and refined the starting site according to the “Predict” step. To further refine the starting site for sequences not starting at M, we took advantage of the reciprocal DELTA-BLAST result of the candidate sequences (details about reciprocal DELTA-BLAST below). Theoretically, if a candidate sequence is similar enough to its top hit in reciprocal DELTA-BLAST, the aligned positions should be close to the full length of the candidate protein, and any residues not aligned on the N terminus are likely artifacts. Following this line of thought, we modified the start site of a candidate sequence to the nearest M before the first aligned position between the sequence and its top hit, if the sequence has more than 10 hits and the aligned length is longer than 60% of the sequence. Start-site refinement was only attempted for sequences from the custom database. Finally, the completeness of the transcriptome assembly was assessed by BUSCO scores v5.1.3 and gene set v10 [Citation50].
Homolog search and annotation
Two rounds of analysis were conducted in order to systematically identify and annotate the ATG homologs, including ATG7, ATG10, ATG12, ATG5, ATG3, ATG8 and ATG4. During the first round, query sequences from Homo sapiens, Saccharomyces cerevisiae, Dictyostelium discoideum and Arabidopsis thaliana were searched against the custom database. During the second round, homologs identified in the first round were queried against both the custom database and NCBI protein and UniProt databases [Citation51,Citation52] downloaded in Jan. 2021. Both rounds used DELTA-BLAST [Citation53] for homology search, at e-value cutoffs of 1e−10 and 1e−6, respectively. In the second round analysis, ATG10 homologs were used as queries for both ATG10 and ATG3, and ATG12 homologs were used as queries for both ATG12 and ATG8.
In each round, candidate sequences identified by DELTA-BLAST [Citation53] were annotated using a combination of conserved domain search [Citation108] and reciprocal DELTA-BLAST [Citation53] against the RefSeq [Citation109] database downloaded in Oct. 2018. Conserved domain search (the online batch tool from https://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi with default options and “standard” result) was able to differentiate ATG10 and ATG3 as ATG3 homologs have additional PF03986 and PF10381 domains on their N and C terminus. However, a recent change in the Pfam [Citation110] domain definitions removed PF03986 and PF10381, making it impossible to separate ATG10 and ATG3 this way. As a work around, we used hmmscan from HMMER v3.3 [Citation54] at e-value cutoff of 1e−6 with an older version of Pfam, v31, in the second round analysis for ATG10 and ATG3. If conserved domain search and reciprocal DELTA-BLAST [Citation53] results disagree, phylogenetic clustering with both Graph Splitting [Citation55] and IQ-TREE [Citation87] was used to resolve the ambiguity. Finally, all annotated sequences were manually inspected following either multiple sequence alignment or alignment with HHsearch [Citation111]. Sequences that are either too short (i.e., covering less than ~50% of well-aligned regions) or without key residues (catalytic cysteines in ATG7, ATG10 and ATG3, the lysine in ATG5 to which ATG12 is conjugated, and catalytic residues in ATG4) were removed. Redundancies in the sequences were removed using CD-HIT [Citation112] at 80% sequence similarity threshold.
For ATG16, special consideration was required because it contains a coiled-coil region and a WD40 domain, both of which exist in many unrelated proteins. In addition, the sequence conservation level of the coiled-coil region in ATG16 homologs can be low in some species. Therefore, additional rounds of analyses were carried out for species with no ATG16 homolog identified from the previous rounds. During the additional rounds, both BLASTP [Citation113] and DETLA-BLAST [Citation53] were used for both the homology search and reciprocal search at e-value cutoff of 0.001 (homology search) and 1e−10 (reciprocal search). Sequences with low e-values, with WD40 domains or aligned to regions outside WD40 domains were prioritized for reciprocal search. Finally, clustering by Graph Splitting was used to remove non-homologs, i.e. those that are not in the region surrounded by green dashed line (Fig. S3). Because the structural maintenance of chromosomes (SMC) proteins are another major family of proteins with coiled-coil regions, eukaryotic SMC proteins from Yoshinaga et al. [Citation114] were included as reference.
Sequence logo
ATG12 and ATG5 sequences were divided into two groups, the normal group and the possibly non-covalent group, and sequence logo was plotted for each group using the ggseqlogo package [Citation115] in R v4.0.3 [Citation116]. The three species with ATG10 but not ATG12 were not included in either group.
Mapping of conservation scores
Multiple sequence alignments of ATG12 and ATG5 were uploaded to the ConSurf server [Citation117] to calculate the conservation scores, which were subsequently manually mapped onto the human ATG12–ATG5-ATG16L1 complex (PDB: 4naw). Surface models color-coded by the conservation levels were plotted using PyMol [Citation118].
Phylogenetic analysis
All ATG3, ATG10, ATG8, and ATG12 sequences identified in this study were included in the phylogenetic analyses. The ATG7 sequences included in Fig. S5 are from the first round analysis. In addition, we collected E1-like, E2-like and ubiquitin-like sequences in the ubiquitin, NEDD8, SUMO1, URM1, and UFM1 systems in three archaeal groups (Asgard, TACK, and Euryarchaeota) and selected eukaryotic species (no E2-like protein for URM1). Sequences were collected with either BLASTP [Citation113] or DELTA-BLAST [Citation53] using the respective human proteins as queries (budding yeast protein for Urm1), and top 15 hits after removing redundancy at 95% were included in the phylogenetic analyses. Phylogenetic trees were reconstructed using both Graph Splitting v2.4 [Citation55] and IQ-TREE v2.1.2 [Citation87]. The sequence similarity matrices produced by Graph Splitting were plotted using the R package igraph (v1.2.11). For IQ-TREE, sequences were aligned using MUSCLE [Citation119] and columns with more than 20% gaps (more than 40% for ATG7) were removed using trimAL [Citation120]. ModelFinder [Citation121] was used to automatically determine the best substitution model to use (LG+R7 for E2-like enzymes, LG+R4 for ubiquitin-like proteins, and VT+R8 for E1-like enzymes), and ten independent runs were conducted. The options used for IQ-TREE are “-alrt 1000 -B 1000 -T 4 -- nmax 5000 --bnni”. Both the ultrafast bootstrap (-B) and SH-aLRT test (-alrt) were used to calculate support values (support values are displayed for main branches in the format of SH-aLRT/Ultrafast bootstrap).
Search of LIR motifs in ATG12, ATG3 and ATG4 homologs
For ATG12, positions corresponding to I111 and F185 in S. cerevisiae were examined for the conservation of I/L/V and F/W/Y residues respectively. For ATG3 and ATG4, LIR motifs were predicted using pLIRm [Citation122]. The LIR motif (WEDL) in Atg3 resides in a mostly fungi-specific loop (Fig. S4), and species with this loop was checked. To annotate if the ATG4 homologs have the two well-conserved LIR motifs, cLIR and APEAR [Citation58,Citation77,Citation78], the corresponding positions in the human ATG4B protein +- 20aa were checked in the multiple sequence alignment.
Supplemental Material
Download Zip (4.2 MB)Acknowledgments
We would like to thank Dr. Yuji Inagaki, University of Tsukuba, for helpful discussions.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Supplementary material
Supplemental data for this article can be accessed here
Additional information
Funding
References
- Mizushima N. The ATG conjugation systems in autophagy. Curr Opin Cell Biol. 2020;63:1–10.
- Maruyama T, Noda NN. Autophagy-regulating protease Atg4: structure, function, regulation and inhibition. J Antibiot (Tokyo). 2018;71(1):72–78.
- Iyer LM, Burroughs AM, Aravind L. The prokaryotic antecedents of the ubiquitin-signaling system and the early evolution of ubiquitin-like β-grasp domains. Genome Biol. 2006;7(7):1–23.
- Maxwell Burroughs A. The natural history of ubiquitin and ubiquitin-related domains. Frontiers in Bioscience. 2012;17(1):1433–1460.
- Burroughs AM, Jaffee M, Iyer LM, et al. Anatomy of the E2 ligase fold: implications for enzymology and evolution of ubiquitin/Ub-like protein conjugation. J Struct Biol. 2008;162(2):205–218.
- Huang B, Jian L, Byström AS. A genome-wide screen identifies genes required for formation of the wobble nucleoside 5-methoxycarbonylmethyl-2-thiouridine in Saccharomyces cerevisiae. Rna. 2008;14(10):2183–2194.
- Schlieker CD, Van Der Veen AG, Damon JR, et al. A functional proteomics approach links the ubiquitin-related modifier Urm1 to a tRNA modification pathway. Proc Natl Acad Sci U S A. 2008;105(47):18255–18260.
- Noma A, Sakaguchi Y, Suzuki T. Mechanistic characterization of the sulfur-relay system for eukaryotic 2-thiouridine biogenesis at tRNA wobble positions. Nucleic Acids Res. 2009;37(4):1335–1352.
- Nakai Y, Nakai M, Hayashi H. Thio-modification of yeast cytosolic tRNA requires a ubiquitin-related system that resembles bacterial sulfur transfer systems. J Biol Chem. 2008;283(41):27469–27476.
- Leidel S, Pedrioli PGA, Bucher T, et al. Ubiquitin-related modifier Urm1 acts as a sulphur carrier in thiolation of eukaryotic transfer RNA. Nature. 2009;458(7235):228–232.
- Furukawa K, Mizushima N, Noda T, et al. A protein conjugation system in yeast with homology to biosynthetic enzyme reaction of prokaryotes. J Biol Chem. 2000;275(11):7462–7465.
- Hochstrasser M. Origin and function of ubiquitin-like proteins. Nature. 2009;458(7237):422–429.
- Liao S, Zhang W, Fan K, et al. Ionic strength-dependent conformations of a ubiquitin-like small archaeal modifier protein (SAMP2) from Haloferax volcanii. Sci Rep. 2013;3(1):1–8.
- Ranjan N, Damberger FF, Sutter M, et al. Solution structure and activation mechanism of ubiquitin-like small archaeal modifier proteins. J Mol Biol. 2011;405(4):1040–1055.
- Xu J, Zhang J, Wang L, et al. Solution structure of Urm1 and its implications for the origin of protein modifiers. Proc Natl Acad Sci U S A. 2006;103(31):11625–11630.
- Yang Z, Chen H, Yang X, et al. A phylogenetic analysis of the ubiquitin superfamily based on sequence and structural information. Mol Biol Rep. 2014;41(9):6083–6088.
- Cajee U-F, Hull R, Ntwasa M. Modification by ubiquitin-like proteins: significance in apoptosis and autophagy pathways. Int J Mol Sci. 2012;13(12):11804–11831.
- Spang A, Saw JH, Jørgensen SL, et al. Complex archaea that bridge the gap between prokaryotes and eukaryotes. Nature. 2015;521(7551):173–179.
- Zaremba-Niedzwiedzka K, Caceres EF, Saw JH, et al. Asgard archaea illuminate the origin of eukaryotic cellular complexity. Nature. 2017;541(7637):353–358.
- Imachi H, Nobu MK, Nakahara N, et al. Isolation of an archaeon at the prokaryote–eukaryote interface. Nature. 2020;577(7791):519–525.
- Nunoura T, Takaki Y, Kakuta J, et al. Insights into the evolution of Archaea and eukaryotic protein modifier systems revealed by the genome of a novel archaeal group. Nucleic Acids Res. 2011;39(8):3204–3223.
- Hennell James R, Caceres EF, Escasinas A, et al. Functional reconstruction of a eukaryotic-like E1/E2/(RING) E3 ubiquitylation cascade from an uncultured archaeon. Nat Commun. 2017;8(1):1–15.
- Fuchs ACD, Maldoner L, Wojtynek M, et al. Rpn11-mediated ubiquitin processing in an ancestral archaeal ubiquitination system. Nat Commun. 2018;9(1):1–12.
- Sakamoto H, Nakada-Tsukui K, Besteiro S. The autophagy machinery in human-parasitic protists; diverse functions for universally conserved proteins. Cells. 2021;10(5):1–19.
- Zhang S, Hama Y, Mizushima N. The evolution of autophagy proteins – diversification in eukaryotes and potential ancestors in prokaryotes. J Cell Sci. 2021;134(13):jcs233742.
- Fischer S, Eichinger L. Dictyostelium discoideum and autophagy – a perfect pair. Int J Dev Biol. 2019;63(8–9–10):485–495.
- Marshall RS, Vierstra RD. Autophagy: the master of bulk and selective recycling. Annu Rev Plant Biol. 2018;69(1):173–208.
- Aslan E, Küçükoglu N, Arslanyolu M. A comparative in-silico analysis of autophagy proteins in ciliates. PeerJ. 2017;5:e2878.
- Kellner R, De la Concepcion JC, Maqbool A, et al. ATG8 expansion: a driver of selective autophagy diversification? Trends Plant Sci. 2017;22(3):204–214.
- Rigden DJ, Michels PA, Ginger ML. Autophagy in protists: examples of secondary loss, lineage-specific innovations, and the conundrum of remodeling a single mitochondrion. Autophagy. 2009;5(6):784–794.
- Seo E, Woo J, Park E, et al. Comparative analyses of ubiquitin-like ATG8 and cysteine protease ATG4 autophagy genes in the plant lineage and cross-kingdom processing of ATG8 by ATG4. Autophagy. 2016;12(11):2054–2068.
- Shemi A, Ben-Dor S, Vardi A. Elucidating the composition and conservation of the autophagy pathway in photosynthetic eukaryotes. Autophagy. 2015;11(4):701–715.
- Wang Q, Liu H, Xu H, et al. Independent losses and duplications of autophagy-related genes in fungal tree of life. Environ Microbiol. 2019;21(1):226–243.
- Pang Y, Yamamoto H, Sakamoto H, et al. Evolution from covalent conjugation to non-covalent interaction in the ubiquitin-like ATG12 system. Nat Struct Mol Biol. 2019;26(4):289–296.
- Adl SM, Bass D, Lane CE, et al. Revisions to the classification, nomenclature, and diversity of eukaryotes. J Eukaryot Microbiol. 2019;66(1):4–119.
- Burki F, Roger AJ, Brown MW, et al. The new tree of eukaryotes. Trends Ecol Evol. 2020;35(1):43–55.
- Moreton J, Izquierdo A, Emes RD. Assembly, assessment, and availability of De novo generated eukaryotic transcriptomes. Front Genet. 2016;6:1–9.
- Brown MW, Heiss AA, Kamikawa R, et al. Phylogenomics places orphan protistan lineages in a novel eukaryotic super-group. Genome Biol Evol. 2018;10(2):427–433.
- Heiss AA, Kolisko M, Ekelund F, et al. Combined morphological and phylogenomic re-examination of malawimonads, a critical taxon for inferring the evolutionary history of eukaryotes. R Soc Open Sci. 2018;5(4):171707.
- Strassert JFH, Jamy M, Mylnikov AP, et al. New phylogenomic analysis of the enigmatic phylum telonemia further resolves the eukaryote tree of life. Mol Biol Evol. 2019;36(4):757–765.
- Burki F, Kaplan M, Tikhonenkov DV, et al. Untangling the early diversification of eukaryotes: a phylogenomic study of the evolutionary origins of centrohelida, haptophyta and cryptista. Proc R Soc B Biol Sci. 2016;283(1823):20152802.
- Cavalier-Smith T, Chao EE, Lewis R. Multiple origins of Heliozoa from flagellate ancestors: new cryptist subphylum Corbihelia, superclass Corbistoma, and monophyly of Haptista, Cryptista, Hacrobia and Chromista. Mol Phylogenet Evol. 2015;93:331–362.
- Cenci U, Sibbald SJ, Curtis BA, et al. Nuclear genome sequence of the plastid-lacking cryptomonad Goniomonas avonlea provides insights into the evolution of secondary plastids. BMC Biol. 2018;16(1):1–23.
- Brown MW, Sharpe SC, Silberman JD, et al. Phylogenomics demonstrates that breviate flagellates are related to opisthokonts and apusomonads. Proc R Soc B Biol Sci. 2013;280(1769):20131755.
- Hampl V, Hug L, Leigh JW, et al. Phylogenomic analyses support the monophyly of Excavata and resolve relationships among eukaryotic “supergroups. Proc Natl Acad Sci U S A. 2009;106(10):3859–3864.
- Lax G, Eglit Y, Eme L, et al. Hemimastigophora is a novel supra-kingdom-level lineage of eukaryotes. Nature. 2018;564(7736):410–414.
- Janouškovec J, Tikhonenkov DV, Burki F, et al. A new lineage of eukaryotes illuminates early mitochondrial genome reduction. Curr Biol. 2017;27(23):3717–3724.
- Grabherr MG, Haas BJ, Yassour M, et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol. 2011;29(7):644–652.
- Haas BJ. TransDecoder; 2015 [cited Oct 222018]. Available from: https://github.com/TransDecoder/TransDecoder
- Seppey M, Manni M, Zdobnov EM. BUSCO: assessing genome assembly and annotation completeness. Methods Mol Biol. 2019;1962:227–245.
- Bateman A, Martin MJ, Orchard S, et al. UniProt: the universal protein knowledgebase in 2021. Nucleic Acids Res. 2021;49(D1):D480–D489.
- Agarwala R, Barrett T, Beck J, et al. Database resources of the national center for biotechnology information. Nucleic Acids Res. 2018;46(D1):D8–D13.
- Boratyn GM, Schäffer AA, Agarwala R, et al. Domain enhanced lookup time accelerated BLAST. Biology Direct. 2012;7(1):1–14.
- Eddy SR. HMMER 3.3; 2019 [cited April15 2020]. Available from: http://hmmer.org
- Matsui M, Iwasaki W. Graph Splitting: a graph-based approach for superfamily-scale phylogenetic tree reconstruction. Syst Biol. 2020;69(2):265–279.
- Kaufmann A, Beier V, Franquelim HG, et al. Molecular mechanism of autophagic membrane-scaffold assembly and disassembly. Cell. 2014;156(3):469–481.
- Yamaguchi M, Noda NN, Nakatogawa H, et al. Autophagy-related Protein 8 (Atg8) family interacting motif in Atg3 Mediates the Atg3-Atg8 Interaction and is crucial for the cytoplasm-to-vacuole targeting pathway. J Biol Chem. 2010;285(38):29599–29607.
- Abreu S, Kriegenburg F, Gómez‐Sánchez R, et al. Conserved Atg8 recognition sites mediate Atg4 association with autophagosomal membranes and Atg8 deconjugation. EMBO Reports. 2017;18(5):765–780.
- Feldmesser E, Rosenwasser S, Vardi A, et al. Improving transcriptome construction in non-model organisms: integrating manual and automated gene definition in Emiliania huxleyi. BMC Genomics. 2014;15:15.
- Albalat R, Cañestro C. Evolution by gene loss. Nat Rev Genet. 2016;17(7):379–391.
- Carosi JM, Nguyen TN, Lazarou M, et al. ATG8ylation of proteins: a way to cope with cell stress? J Cell Biol. 2021;220(11):1–4.
- Nguyen TN, Padman BS, Zellner S, et al. ATG4 family proteins drive phagophore growth independently of the LC3/GABARAP lipidation system. Mol Cell. 2021;81(9):2013–2030.
- Agrotis A, Pengo N, Burden JJ, et al. Redundancy of human ATG4 protease isoforms in autophagy and LC3/GABARAP processing revealed in cells. Autophagy. 2019;15(6):976–997.
- Hanada T, Noda NN, Satomi Y, et al. The Atg12-Atg5 conjugate has a novel E3-like activity for protein lipidation in autophagy. J Biol Chem. 2007;282(52):37298–37302.
- Zheng Y, Qiu Y, Grace CRR, et al. A switch element in the autophagy E2 Atg3 mediates allosteric regulation across the lipidation cascade. Nat Commun. 2019;10(1):1–14.
- Otomo C, Metlagel Z, Takaesu G, et al. Structure of the human ATG12 ATG5 conjugate required for LC3 lipidation in autophagy. Nat Struct Mol Biol. 2013;20(1):59–66.
- Noda NN, Fujioka Y, Hanada T, et al. Structure of the Atg12-Atg5 conjugate reveals a platform for stimulating Atg8-PE conjugation. EMBO Rep. 2013;14(2):206–211.
- Reggiori F, Monastyrska I, Verheije MH, et al. Coronaviruses hijack the LC3-I-positive EDEMosomes, ER-derived vesicles exporting short-lived ERAD regulators, for replication. Cell Host Microbe. 2010;7(6):500–508.
- Mikawa T, Kanoh J, Ishikawa F. Fission yeast Vps1 and Atg8 contribute to oxidative stress resistance. Genes Cells. 2010;15(3):229–242.
- Tamura N, Oku M, Sakai Y. Atg8 regulates vacuolar membrane dynamics in a lipidation-independent manner in Pichia pastoris. J Cell Sci. 2010;123(23):4107–4116.
- Liu X-M, Yamasaki A, Du X-M, et al. Lipidation-independent vacuolar functions of atg8 rely on its noncanonical interaction with a vacuole membrane protein. Elife. 2018;7:1–21.
- Ishii A, Kurokawa K, Hotta M, et al. Role of Atg8 in the regulation of vacuolar membrane invagination. Sci Rep. 2019;9(1):1–11.
- Jipa A, Vedelek V, Merényi Z, et al. Analysis of Drosophila Atg8 proteins reveals multiple lipidation-independent roles. Autophagy. 2020;17(9):2565–2575.
- Bhattacharya D, Qiu H, Lee JM, et al. When less is more: red algae as models for studying gene loss and genome evolution in Eukaryotes. CRC Crit Rev Plant Sci. 2018;37(1):81–99.
- Pipaliya SV, Santos R, Salas-Leiva D, et al. Unexpected organellar locations of ESCRT machinery in Giardia intestinalis and complex evolutionary dynamics spanning the transition to parasitism in the lineage Fornicata. BMC Biol. 2021;19(1):19.
- Wadi L, Reinke AW. Evolution of microsporidia: an extremely successful group of eukaryotic intracellular parasites. PLoS Pathog. 2020;16(2):e1008276.
- Skytte Rasmussen M, Mouilleron S, Kumar Shrestha B, et al. ATG4B contains a C-terminal LIR motif important for binding and efficient cleavage of mammalian orthologs of yeast Atg8. Autophagy. 2017;13(5):834–853.
- Satoo K, Noda NN, Kumeta H, et al. The structure of Atg4B–LC3 complex reveals the mechanism of LC3 processing and delipidation during autophagy. EMBO J. 2009;28(9):1341–1350.
- Kong-Hap MA, Mouammine A, Daher W, et al. Regulation of ATG8 membrane association by ATG4 in the parasitic protist Toxoplasma gondii. Autophagy. 2013;9(9):1334–1348.
- Liu ML, Yao MC. Role of ATG8 and Autophagy in programmed nuclear degradation in Tetrahymena thermophila. Eukaryot Cell. 2012;11(4):494–506.
- Fujita N, Itoh T, Omori H, et al. The Atg16L complex specifies the site of LC3 lipidation for membrane biogenesis in autophagy. Mol Biol Cell. 2008;19(5):2092–2100.
- Matsushita M, Suzuki NN, Obara K, et al. Structure of Atg5 Atg16, a Complex Essential for Autophagy. J Biol Chem. 2007;282(9):6763–6772.
- Popelka H, Reinhart EF, Metur SP, et al. Membrane Binding and Homodimerization of Atg16 Via Two Distinct Protein Regions is Essential for Autophagy in Yeast. J Mol Biol. 2021;433(5):166809.
- Nishimura T, Kaizuka T, Cadwell K, et al. FIP200 regulates targeting of Atg16L1 to the isolation membrane. EMBO Rep. 2013;14(3):284–291.
- Gammoh N, Florey O, Overholtzer M, et al. Interaction between FIP200 and ATG16L1 distinguishes ULK1 complex-dependent and-independent autophagy. Nat Struct Mol Biol. 2013;20(2):144–149.
- Schatz D, Shemi A, Rosenwasser S, et al. Hijacking of an autophagy-like process is critical for the life cycle of a DNA virus infecting oceanic algal blooms. New Phytol. 2014;204(4):854–863.
- Nguyen LT, Schmidt HA, Von Haeseler A, et al. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol. 2015;32(1):268–274.
- Koonin EV, Yutin N. The dispersed archaeal eukaryome and the complex archaeal ancestor of eukaryotes. Cold Spring Harb Perspect Biol. 2014;6(4):a016188.
- Fujioka Y, Noda NN, Fujii K, et al. In vitro reconstitution of plant Atg8 and Atg12 conjugation systems essential for autophagy. J Biol Chem. 2008;283(4):1921–1928.
- Arisue N, Hashimoto T. Phylogeny and evolution of apicoplasts and apicomplexan parasites. Parasitol Int. 2015;64(3):254–259.
- Cavalier-Smith T, Fiore-Donno AM, Chao E, et al. Multigene phylogeny resolves deep branching of Amoebozoa. Mol Phylogenet Evol. 2015;83:293–304.
- Lemieux C, Otis C, Turmel M. Chloroplast phylogenomic analysis resolves deep-level relationships within the green algal class Trebouxiophyceae. BMC Evol Biol. 2014;14(1):1–15.
- Turmel M, De Cambiaire J-C, Otis C, et al. Distinctive architecture of the chloroplast genome in the chlorodendrophycean green algae scherffelia dubia and tetraselmis sp. CCMP 881. PLoS One. 2016;11(2):1–22.
- Yang J, Harding T, Kamikawa R, et al. Mitochondrial genome evolution and a novel RNA editing system in deep-branching heteroloboseids. Genome Biol Evol. 2017;9(5):1161–1174.
- Torruella G, De Mendoza A, Grau-Bové X, et al. Phylogenomics reveals convergent evolution of lifestyles in close relatives of animals and fungi. Curr Biol. 2015;25(18):2404–2410.
- Torruella G, Grau-Bové X, Moreira D, et al. Global transcriptome analysis of the aphelid Paraphelidium tribonemae supports the phagotrophic origin of fungi. Commun Biol. 2018;1(1):1–11.
- Karpov SA, Mamkaeva MA, Aleoshin VV, et al. Morphology, phylogeny, and ecology of the aphelids (Aphelidea, Opisthokonta) and proposal for the new superphylum Opisthosporidia. Front Microbiol. 2014;5:1–11.
- Carr M, Leadbeater BSC, Hassan R, et al. Molecular phylogeny of choanoflagellates, the sister group to Metazoa. Proc Natl Acad Sci U S A. 2008;105(43):16641–16646.
- Sato M, Arita M, Kawashima T. Uncovering ecdysozoa-specific sphingomyelin synthase by phylogenetic analysis of metazoan sequences. Zoolog Sci. 2019;36(4):316–321.
- Karnkowska A, Treitli SC, Brzoň O, et al. The oxymonad genome displays canonical eukaryotic complexity in the absence of a mitochondrion. Mol Biol Evol. 2019;36(10):2292–2312.
- Burki F, Kudryavtsev A, Matz MV, et al. Evolution of Rhizaria: new insights from phylogenomic analysis of uncultivated protists. BMC Evol Biol. 2010;10(1):1–18.
- Sierra R, Matz MV, Aglyamova G, et al. Deep relationships of Rhizaria revealed by phylogenomics: a farewell to Haeckel’s Radiolaria. Mol Phylogenet Evol. 2013;67:53–59.
- Thakur R, Shiratori T, Ichiro IK. Taxon-rich multigene phylogenetic analyses resolve the phylogenetic relationship among deep-branching stramenopiles. Protist. 2019;170(5):125682.
- Muñoz-Gómez SA, Mejía-Franco FG, Durnin K, et al. The new red algal subphylum proteorhodophytina comprises the largest and most divergent plastid genomes known. Curr Biol. 2017;27(11):1677–1684.
- Leinonen R, Sugawara H, Shumway M. The sequence read archive. Nucleic Acids Res. 2011;39(Database):2010–2012.
- Keeling PJ, Burki F, Wilcox HM, et al. The Marine Microbial Eukaryote Transcriptome Sequencing Project (MMETSP): illuminating the functional diversity of eukaryotic life in the oceans through transcriptome sequencing. PLoS Biol. 2014;12(6):e1001889.
- Chen S, Zhou Y, Chen Y, et al. Fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics. 2018;34(17):i884–i890.
- Lu S, Wang J, Chitsaz F, et al. CDD/SPARCLE: the conserved domain database in 2020. Nucleic Acids Res. 2020;48(D1):D265–D268.
- O’Leary NA, Wright MW, Brister JR, et al. Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 2016;44(D1):D733–D745.
- El-Gebali S, Mistry J, Bateman A, et al. The Pfam protein families database in 2019. Nucleic Acids Res. 2019;47(D1):D427–D432.
- Steinegger M, Meier M, Mirdita M, et al. HH-suite3 for fast remote homology detection and deep protein annotation. BMC Bioinformatics. 2019;20(1):1–15.
- Fu L, Niu B, Zhu Z, et al. CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics. 2012;28(23):3150–3152.
- Altschul SF, Gish W, Miller W, et al. Basic local alignment search tool. J Mol Biol. 1990;215(3):403–410.
- Yoshinaga M, Inagaki Y. Ubiquity and Origins of Structural Maintenance of Chromosomes (SMC) Proteins in Eukaryotes. Genome Biol Evol. 2021;13(12):1–17.
- Wagih O. Ggseqlogo: a versatile R package for drawing sequence logos. Bioinformatics. 2017;33(22):3645–3647.
- R Core Team. R: a language and environment for statistical computing. Vienna Austria: R Foundation for Statistical Computing; 2020. https://www.r-project.org
- Ashkenazy H, Abadi S, Martz E, et al. ConSurf 2016: an improved methodology to estimate and visualize evolutionary conservation in macromolecules. Nucleic Acids Res. 2016;44(W1):W344–W350.
- The PyMOL Molecular Graphics System, Version 2.3.4. Schrödinger, LLC; 2015.
- Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32(5):1792–1797.
- Capella-Gutiérrez S, Silla-Martínez JM, Gabaldón T. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics. 2009;25(15):1972–1973.
- Kalyaanamoorthy S, Minh BQ, Wong TKF, et al. ModelFinder: fast model selection for accurate phylogenetic estimates. Nat Methods. 2017;14(6):587–589.
- Han Z, Zhang W, Ning W, et al. Model-based analysis uncovers mutations altering autophagy selectivity in human cancer. Nat Commun. 2021;12(1):12.