
ABSTRACT
Although the involvement of macroautophagy/autophagy in hepatitis B virus (HBV) infection has become clearer recently, whether selective autophagy plays an important role in suppressing HBV remains uncertain. We recently found that LGALS9 (galectin 9) is an interferon (IFN)-inducible protein involved in the suppression of HBV replication. Expression of LGALS9 in HBV-infected cells causes the formation of cytoplasmic puncta that degrade the HBV core protein (HBc) in conjunction with RSAD2/viperin, another IFN-inducible protein. LGALS9 binds to HBc via RSAD2 and promotes the autoubiquitination of RNF13 (ring finger protein 13) to recruit SQSTM1/p62, resulting in the formation of LC3-positive autophagosomes that degrade HBc. Both LGALS9 and RSAD2 are encoded by IFN-stimulated genes that act synergistically to induce HBc proteolysis in HBV-infected hepatocytes in an IFN-dependent manner. These results reveal a crosstalk mechanism between the innate immune system and selective autophagy during viral infection.
Text
Hepatitis B virus (HBV) is a blood-borne pathogen that causes chronic infection. Currently, an estimated 350 million individuals worldwide have chronic HBV infection (CHB) and are at a risk of progression to cirrhosis and hepatocellular carcinoma. Although the currently available treatment options for HBV can prevent progression to cirrhosis and hepatocellular carcinoma, none of them can eliminate the HBV genome in chronically infected cells.
One of the most potent treatment options for CHB is type-I interferon (IFN) administration. As IFNs are the principal agents of innate immunity that cause viral clearance, they could possibly eliminate the chronic reservoir of HBV. In addition to innate immunity-mediated viral clearance by IFNs, xenophagy, a form of selective autophagy, has been demonstrated to eliminate virus-infected cells. Whether these two mechanisms act independently or cooperatively is currently unknown. Therefore, we aimed to determine if a connection exists between IFN and xenophagy-mediated clearance of HBV in infected cells, in pursuance of a controlled and efficient elimination of HBV reservoirs.
HBV encodes four proteins, namely HBc, HBx, Pol, and HBs. HBc is the structural component of the viral nucleocapsid and plays a pivotal role in the synthesis of covalently closed circular DNA (cccDNA) during the viral life cycle. As HBc exacerbates hepatic inflammation in CHB, we focused our research on the involvement of this viral molecule in innate immunity and xenophagy.
In our recent work [Citation1], we generated a cDNA library comprising 130 interferon stimulatory genes (ISGs) whose expression was specifically induced by type-I IFN in human hepatocytes. Each member of the ISG family was co-transfected with HBc in living cells, and the bioluminescence resonance energy transfer (BRET) method was used to screen for ISG proteins in the proximity of HBc. Results showed that 10 different ISG proteins interact with HBc, of which LGALS9 (galectin 9) markedly reduces HBV release. In the absence of LGALS9, HBc is diffusely scattered in the cytoplasm and to a lesser extent in the nucleus; however, upon LGALS9 expression, HBc localizes to the cytoplasmic puncta with LGALS9 in proximity. This was surprising, as LGALS9 is known to solely bind to carbohydrate moieties of glycosylated host proteins and not to the glycosylated residues of viral proteins such as HBc. In vitro affinity-isolation analysis using recombinant proteins demonstrated that the direct binding of LGALS9 to HBc is unlikely, suggesting the existence of other molecule(s) affecting the binding between HBc and LGALS9. Using the BRET screening associated with a bioinformatics database, we identified another interferon-inducible factor, RSAD2/viperin, as the linker molecule that mediates LGALS9-HBc binding. Furthermore, the autophagosome marker LC3 is localized at the puncta containing the LGALS9-RSAD2-HBc complex. We also observed that LGALS9 can bind to the ubiquitin ligase RNF13. This binding causes autoubiquitination of RNF13, leading to the recruitment of SQSTM1, one of the receptors for selective autophagy and resulting in the formation of LC3-positive autophagosomes ().
Figure 1. Illustration of the mechanism by which LGALS9 recruits multiple factors and induces selective autophagy of the HBV core protein.
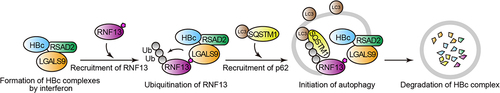
Because LGALS9 was found to be an IFN-inducible protein expressed through ISGs, we next examined whether type-I IFN can induce HBc accumulation in LC3-positive puncta. IFNB/IFN-β treatment increases the colocalization of HBc and LC3 in the cytoplasm, which is abolished by depletion of LGALS9. Taken together, these results suggest that IFN-inducible proteins (LGALS9 and RSAD2) act in coalition with molecules of the autophagic pathway (RNF13 and SQSTM1) to transport HBc to LC3-positive autophagic puncta, leading to autophagy-mediated degradation of the viral protein.
Proteins of the galectin family are involved in various immunological processes, including immune cell activation, cytokine production, inflammation, and cancer immunity. Existing literature suggests that the galectin family acts as an intracellular sensor for organelle membrane damage by identifying host proteins displaying carbohydrate moieties released during the damage. However, we found a novel role for LGALS9 in xenophagy via indirect binding to viral proteins. The results reveal a broad perspective in which the complex events that cause serial cooperation of autophagic pathway molecules are directly regulated by innate immunity, and IFNs act as an ON-OFF switch in this process. Our findings might provide valuable insights into a new therapeutic strategy that can efficiently eliminate HBV reservoirs and reduce the burden of chronic HBV infection by utilizing the autophagic pathway.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
Reference
- Miyakawa K, Nishi M, Ogawa M, et al. Galectin-9 restricts hepatitis B virus replication via p62/SQSTM1-mediated selective autophagy of viral core proteins. Nat Commun. 2022;13(1):531.