
ABSTRACT
Neurons are highly polarized and functionally compartmentalized cells. Under basal conditions, the biogenesis of autophagic vesicles (AVs) was previously shown to take place in the axon tip. As the sequestration of autophagic cargo occurs during the formation of nascent AVs, this would mean that only axonal proteins can be degraded via macroautophagy/autophagy, unless AV biogenesis can also take place on demand, in other neuronal compartments. Our work shows that indeed, activation of NMDA or group I metabotropic glutamate receptors during long-term synaptic depression (LTD) triggers the biogenesis of AVs locally in dendrites. Under these conditions, nascent dendritic AVs are required for synaptic plasticity, as they sequester postsynaptic proteins, whose removal from the postsynapse is necessary for LTD.
Main text
Most neurons have complex morphologies, comprised of long and branching processes that are functionally compartmentalized: Axons that contain the presynaptic terminals at their tips, and dendrites, often spiny, harboring postsynaptic specializations. Where are new autophagic vesicles (AVs) produced in this highly polarized cytoarchitecture? Previous work demonstrated that under basal conditions, AV biogenesis occurs almost exclusively at the axon tip, placing it in the vicinity of presynaptic terminals. This is in sharp contrast to other cell types, where AV biogenesis occurs indiscriminately throughout the cytoplasm. In line with this finding, several studies have identified presynaptic interactors of autophagic proteins and autophagy has been proposed to regulate the degradation and release of synaptic vesicles in some neuronal types. Notably, autophagy is not the only cellular process, and AVs not the only organelles, that show a non-ubiquitous distribution in neurons. For example, lysosomes are concentrated in the neuronal soma, meaning that most nascent axonal AVs need to travel unidirectionally to the soma to deliver their cargo.
However, this skewed distribution would imply that postsynaptic specializations, mostly concentrated in dendrites, are not functionally subserved by the autophagic machinery. Yet, paradoxically, emerging work has highlighted the requirement of autophagy for key behaviors, such as memory formation and erasure, that are underlined by long-term synaptic plasticity, namely cellular mechanisms which are mainly expressed postsynaptically. Similarly, autophagy is necessary for the developmental pruning of dendritic spines, a process that is essential for the fine-tuning of synaptic connectivity during early postnatal life, and whose failure leads to behavioral deficits associated with autism spectrum disorders.
We suspected that AV biogenesis may be triggered on demand in dendrites when the sequestration and degradation of postsynaptic proteins is needed. In search of biologically relevant paradigms, we focused on the process of long-term depression (LTD) for several reasons: First, LTD is a fundamental form of long-term plasticity underlying cognitive flexibility, memory erasure and learning; some of these behaviors are impaired when autophagy is perturbed. Second, at the cellular level LTD is facilitated by the removal of glutamate AMPA receptors from the postsynaptic density and their subsequent degradation by mechanisms that remain poorly characterized, often leading to the shrinkage or elimination of entire dendritic spines. Finally, LTD-like mechanisms facilitate the developmental pruning of dendritic spines, a process which, as already mentioned, requires autophagy. For these reasons, we speculated that there may be an interplay between LTD and AV biogenesis and set out to investigate it.
Two major forms of LTD co-exist in the brain, mediated either by NMDA or group I metabotropic glutamate receptors (abbreviated as NMDAR and mGluR, respectively). Kallergi, Daskalaki and colleagues demonstrated that both NMDAR- and mGluR-LTD trigger the rapid assembly of ULK1 complexes, and the sequential formation of phagophores and AVs in postsynaptic dendrites, both in vitro and in acute hippocampal slices [Citation1]. This conclusion was supported by imaging of endogenous dendritic puncta positive for the four ULK1-complex components and of endogenous LC3-positve puncta, as well as by electron tomography of LTD-activated postsynaptic spines and dendrites in the hippocampus.
These findings invited the speculation that dendritic autophagy is not an accidental bystander of LTD, but, instead, a mechanism specifically recruited on demand for the degradation of postsynaptic components and the expression of LTD. To investigate this, we examined the requirement of autophagy for LTD induction in the well-established hippocampal CA3-CA1 circuit, using both genetic and pharmacological tools to impair autophagy. These experiments confirmed that both NMDAR- and mGluR-LTD fail to be induced in the hippocampus when a) the ULK1 kinase activity is acutely inhibited during the induction phase, b) Atg5 is ablated in pyramidal neurons across the forebrain or c) Atg5 is knocked down specifically in postsynaptic CA1 neurons of the hippocampus. The consequences of failed LTD are behaviorally manifested in mice with conditional ablation of Atg5 in pyramidal neurons as altered responses in a cognitive flexibility test. This observation adds to other LTD-related behaviors, such as memory formation and erasure, which, as already mentioned, also require intact autophagy.
We postulated that dendritic AVs facilitate LTD via their canonical function in protein degradation. To examine if this is the case, AVs were isolated from hippocampal slices before and after LTD induction and their content was compared by quantitative proteomic analyses. Despite having analyzed not only dendritic AVs, we found a significant enrichment of postsynaptic proteins in the AV content after LTD. These included the GRIA1/GluA1 subunit of AMPARs and its key scaffold protein DLG4/PSD95, whose autophagic degradation was confirmed by their accumulation after LTD when lysosomal acidification or AV biogenesis were prevented.
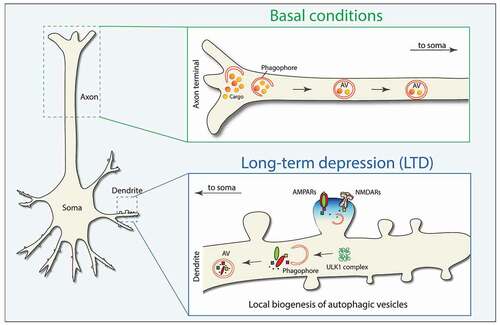
As summarized in , we have revealed that a specific synaptic process, LTD, triggers the on-demand biogenesis of AVs in dendrites to facilitate the degradation of precise postsynaptic proteins, conducive to facilitating a sustained depression of synaptic strength. These findings will hopefully lead the way to revisiting the interplay between autophagy and other forms of synaptic plasticity and its implications for normal behavior and relevant disease-related phenotypes.
Figure 1. Schematic representation of basal AV biogenesis in the axonal tip (top panel) and on demand AV biogenesis in dendrites (bottom panel) during synaptic plasticity.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
Reference
- Kallergi E, Daskalaki AD, Kolaxi A, et al. Dendritic autophagy degrades postsynaptic proteins and is required for long-term synaptic depression in mice. Nat Commun. 2022;13(1):680.