
ABSTRACT
SQSTM1/p62 is a selective macroautophagy/autophagy receptor that drives ubiquitinated cargos toward the lysosome for degradation, and also a stress-induced scaffold protein that helps cells to cope with oxidative stress through sequestrating KEAP1 and subsequent activation of the NFE2L2/NRF2 antioxidant pathway. Accumulating evidence implicates SQSTM1 dysregulation in the induction of multiple oncogenic transformations in vivo. SPOP (speckle type BTB/POZ protein), an E3 ubiquitin ligase adaptor, is the most frequently mutated gene in prostate cancer (Pca), but the molecular mechanisms underlying how SPOP mutations contribute to PCa tumorigenesis are still largely unknown. In a recent study, we describe a new role for SPOP as a negative regulator of autophagy and NFE2L2 pathway activation. SPOP binds and induces the non-degradative ubiquitination of SQSTM1 at Lys420. This post-translational modification decreases SQSTM1 body formation, liquid phase condensation, dimerization, and ubiquitin-binding capacity, thereby suppressing SQSTM1-dependent autophagy, KEAP1 sequestration, and NFE2L2 activation. Notably, PCa-associated SPOP mutants lose the capacity to ubiquitinate SQSTM1 and instead enhance autophagy and the antioxidant response in a dominant-negative manner. Thus, our findings indicate the critical roles of autophagy and NFE2L2 pathway activation in PCa tumorigenesis by oncogenic SPOP mutations.
Identification and functional characterization of the genetic alterations that initiate human cancers and contribute to its progression are necessary for effective cancer prevention and treatment. SPOP (speckle type BTB/POZ protein) is one of the substrate-binding adaptors of the CUL3 (cullin 3)-RING E3 ubiquitin ligase complexes (CRL3). Accumulating evidence revealed that heterozygous SPOP mutations are recurrently found in 5–15% of two hormone-related cancers, prostate cancer (PCa) and endometrial cancer (EC), but such mutations are relatively low in cancers of other tissue types. The majority of PCa or EC-associated SPOP mutations are within the substrate-binding MATH domain, suggesting that there is a change in binding affinity between SPOP and its interacting partners. We and others have identified multiple oncoproteins, such as NCOA3/SRC-3, AR, ESR1/ERα, DEK, BRD2, BRD3 and BRD4, are authentic proteolytic substrates of the CRL3-SPOP complex. Moreover, the CRL3-SPOP complex exerts its tumor-suppressive capacity by promoting the non-degradative ubiquitination of several substrates, such as MACROH2A, INF2, MYD88 and HIPK2. Nonetheless, how SPOP mutations contribute to the pathogenesis and progression of human cancers remains poorly understood.
SQSTM1/p62 is a one of the selective autophagy receptors that cross-link poly-ubiquitinated cargos and autophagosomal Atg8-family proteins to deliver them for lysosomal degradation, as well as in a wide range of signaling activities as part of the cellular response to oxidative stress, nutrient sensing, and inflammation. SQSTM1 sequesters the CRL3 adaptor KEAP1 (kelch like ECH associated protein 1), thereby leading to increased stabilization and activity of NFE2L2/NRF2, a key transcription factor in reprogramming oxidative stress responses. The aberrant activation of the SQSTM1-KEAP1-NFE2L2 pathway, which is frequently observed in various human cancers, protects cells against oxidative stress-induced cell death and leads to tumorigenesis in vivo. Although the pathophysiological importance of SQSTM1 is well-recognized, the dynamic regulation of SQSTM1 activity under basal and stress conditions is not fully understood.
In a recent study, we described a new role for SPOP as a negative regulator of autophagy and the NFE2L2 antioxidant pathway in a SQSTM1-dependent manner [Citation1]. To identify SPOP substrates whose dysregulation play roles in SPOP mutation-driven PCa tumorigenesis, we performed an affinity purification followed by mass spectrometry to identify new SPOP-interacting partners, and SQSTM1 ranked high on the interaction hit list. We confirmed that these two proteins interact with each other, and the MATH domain of SPOP binds SQSTM1 by recognizing a conserved SPOP-binding consensus motif (SBC) in SQSTM1. No obvious protein changes of SPOP and its substrates (BRD4, DEK, and CAPRIN1) are observed in SQSTM1, ATG3, ATG5, and ATG7 knockout cells. These results indicated that SPOP is not an autophagic substate and SQSTM1 has no impact on SPOP-mediated substrate degradation, which is distinct from its roles toward KEAP1. We then explored an alternative possibility that SPOP regulates SQSTM1 function. In contrast to the degradative feature toward its proteolytic substrates, SPOP has no impact on SQSTM1 protein level but promotes the non-degradative K6, K27, and K29 mixed-linkage polyubiquitination of SQSTM1, and K420 within the UBA domain of SQSTM1 serves as the predominant ubiquitin attachment site. In accordance with previous studies, PCa-associated SPOP mutants show impaired SQSTM1-binding capacity and function as dominant-negative variants to downregulate SQSTM1 ubiquitination instead. Given that SQSTM1 interacts with SPOP in an SBC motif-dependent manner, we further demonstrated that a subset of cancer-associated mutations that occur in the SBC motif of SQSTM1 attenuate the SPOP-SQSTM1 interaction and permit SQSTM1 to evade SPOP-mediated ubiquitination.
Liquid-liquid phase separation (LLPS) promotes the formation of membraneless condensates that mediate diverse cellular functions. Previous studies suggest that cytoplasmic SQSTM1 bodies are subject to LLPS and have viscous liquid-like properties, which serve as platforms for autophagosome formation and NFE2L2 activation. The UBA domain-mediated polyUb chain binding induces SQSTM1 LLPS. However, the regulatory mechanisms underlying this process are still poorly understood. our findings indicated that wild-type SPOP, but not the PCa-associated mutants, are colocalized with SQSTM1 as cytoplasmic puncta. SPOP-mediated SQSTM1 ubiquitination negatively regulates SQSTM1 LLPS, thereby reducing its dimerization and ubiquitin-binding capacity. Further, we showed that wild-type SPOP restricts rapamycin-induced autophagy, while PCa-associated mutants instead enhance autophagy in a dominant-negative fashion. Moreover, the negative impact of SPOP on SQSTM1 is relieved when autophagy is initiated, suggesting this process can be dynamically regulated by external stimuli.
SQSTM1 plays critical roles in the NFE2L2-activated antioxidant response by sequestering KEAP1 into aggregates, resulting in the inhibition of KEAP1-mediated NFE2L2 degradation. Reciprocally, KEAP1 facilitates the SQSTM1-mediated ubiquitin aggregate clearance via autophagy by ubiquitinating SQSTM1 at Lys420. We found that SPOP negatively regulates SQSTM1-mediated KEAP1 sequestration and this effect is strictly dependent on the SPOP-SQSTM1 interaction and SPOP-mediated SQSTM1 ubiquitination. The fact that SPOP-mediated SQSTM1 ubiquitination negatively regulates SQSTM1 body formation is distinct from KEAP1-mediated SQSTM1 ubiquitination, which promotes SQSTM1 body formation, although these two events modify the same Lys420 residues in SQSTM1. It is possible that the different linkage types of polyUb chain catalyzed by CRL3-SPOP or CRL3-KEAP1 on SQSTM1 determine the distinct SQSTM1 fates. Finally, we showed that SPOP-deficient or PCa-associated SPOP mutant-overexpressing cells display an enhanced antioxidant response and reduced cell death in response to oxidative stress in a NFE2L2-dependent manner.
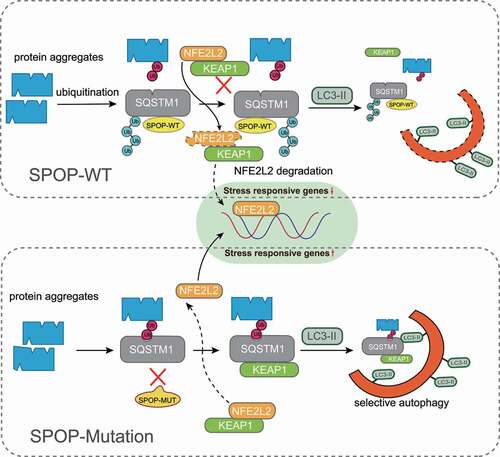
Collectively, our study demonstrated that PCa-associated SPOP mutations enhance autophagy and NFE2L2 activation by directly modulating SQSTM1 LLPS and ubiquitination (). Elucidation of this oncogenic pathway will guide targeted therapy toward SPOP-mutated cancers.
Figure 1. A Schematic diagram depicts a model where both autophagy and the NFE2L2 pathway are abnormally elevated in SPOP-mutated cancer cells.
Disclosure statement
All authors have no conflicts of interest to declare.
Additional information
Funding
Reference
- Shi Q, Jin X, Zhang P, et al. SPOP mutations promote p62/SQSTM1-dependent autophagy and Nrf2 activation in prostate cancer. Cell Death Differ. 2022 Jan 6. DOI:https://doi.org/10.1038/s41418-021-00913-w.