
ABSTRACT
Enterovirus D68 (EV-D68) is a respiratory pathogen associated with acute flaccid myelitis, a childhood paralysis disease. No approved vaccine or antiviral treatment exists against EV-D68. Infection with this virus induces the formation of autophagosomes to enhance its replication but blocks the downstream autophagosome- lysosome fusion steps. Here, we examined the impact of autophagy induction through starvation, either before (starvation before infection, SBI) or after (starvation after infection, SAI) EV-D68 infection. We showed that SAI, but not SBI, attenuated EV-D68 replication in multiple cell lines and abrogated the viral-mediated cleavage of host autophagic flux-related proteins. Furthermore, SAI induced autophagic flux during EV-D68 replication and prevented production of virus-induced membranes, which are required for picornavirus replication. Pharmacological inhibition of autophagic flux during SAI did not rescue EV-D68 titers. SAI had the same effect in multiple cell types, and restricted the replication of several medically relevant picornaviruses. Our results highlight the significance of autophagosomes for picornavirus replication and identify SAI as an attractive broad-spectrum anti-picornavirus strategy.
Abbreviations: BAF: bafilomycin A1; CCCP: carbonyl cyanide m-chlorophenylhydrazone; CQ: chloroquine; CVB3: coxsackievirus B3; EV-D68: enterovirus D68; hpi: hour post-infection; MAP1LC3/LC3: microtubule associated protein 1 light chain 3; MOI: multiplicity of infection; NSP2B: nonstructural protein 2B; PV: poliovirus; RES: resveratrol; RV14: rhinovirus 14; SAI: starvation after infection; SBI: starvation before infection; SNAP29: synaptosome associated protein 29; SQSTM1/p62: sequestosome 1; TFEB: transcription factor EB.
KEYWORDS:
Introduction
First discovered in 1962, the incidence of enterovirus D68 (EV-D68) infection has been on the rise over the past decade. Since its discovery, EV-D68 has caused clusters of acute respiratory illness in multiple countries, and recent shreds of evidence implicate its infection with a rare polio-like neurological disease, acute flaccid myelitis, in children making EV-D68 a respiratory pathogen of public health importance [Citation1–3]. Currently, there are no effective antivirals or prophylactic vaccines against EV-D68 infection. Therefore, understanding how EV-D68 interacts with the machinery of its host cells could open avenues for therapeutic interventions against the enterovirus.
EV-D68 belongs to the Piconarviridae family of viruses, which includes important human and animal viruses such as poliovirus, coxsackievirus B3, enterovirus A71, hepatitis A virus, and Seneca Valley virus, among others [Citation4,Citation5]. The viral genome is 7500 nucleotides long and encodes a single polyprotein that is processed by viral proteases to four structural proteins (VP1, VP2, VP3, and VP4), which enwrap the viral RNA and are responsible for viral binding and entry into the susceptible cells, and seven nonstructural proteins (2A, 2B, 2C, 3A, 3B, 3C, and 3D) which among other functions, rearrange cellular membranes and are responsible for viral replication. Picornavirus replication is thought to occur on single and double-membrane vesicles, which resemble autophagosomes [Citation6]. Indeed, multiple lines of evidence have shown that picornaviruses utilize autophagosomes for RNA replication [Citation7–9].
Macroautophagy (hereafter autophagy) is a conserved catabolic process that targets long-lived proteins and damaged cytoplasmic organelles to the lysosomes for degradation. The cellular process can also target microorganisms, including bacteria and viruses, for degradation through xenophagy, forming an essential part of the immune response [Citation10]. Autophagy can be induced by several stress stimuli, including amino acid starvation. Autophagy begins with the formation of the phagophore, which expands and elongates through two ubiquitin-like conjugation systems: the conjugation of ATG5, ATG12, and ATG16L1; and the addition of phosphatidylethanolamine (PE) to MAP1LC3B/LC3B (microtubule associated protein 1 light chain 3 beta) to form the autophagosome, which then fuses with the lysosomes to degrade the cargo [Citation11]. The lipidation of LC3B (conversion of soluble LC3B-I to the membrane-bound LC3B-II) and the degradation of the receptor protein, SQSTM1, is widely used to monitor autophagy [Citation12].
Although autophagy has long been known for its antimicrobial activity, several RNA viruses, including picornaviruses, are known to subvert and even repurpose the cellular process to benefit their replication [Citation13]. While autophagy serves as a proviral mechanism in the life cycle of many picornaviruses, these viruses tend to block autophagic flux [Citation14], which is thought to both maximize viral replication by providing more autophagosomes and prevent degradation of virions or viral proteins. Indeed, we and others have recently shown that EV-D68 employs multiple strategies, including cleavage of both SQSTM1 and SNAP29 (synaptosome associated protein 29), a SNARE protein which is critical for autophagosome-lysosome fusion, to attenuate autophagic flux [Citation15,Citation16]. Given the importance of autophagosomes in the picornavirus lifecycles and the multiple strategies employed by these infectious agents to block flux, we hypothesized that inducing autophagic flux early in EV-D68 infection could restrict viral replication. Here, we demonstrated that Starvation After Infection (SAI) reduces EV-D68 replication in multiple cell lines. We suggest that this is because expediting the turnover of autophagosomes depletes the cell of resources to build replication membranes, which are crucial for viral RNA replication. Furthermore, we show that the anti-picornavirus activity of our SAI protocol does not implicate degrading viral components in the autolysosomes, as pharmacological inhibition of autophagic flux did not rescue the viral titer. Our results highlight the importance of autophagosome development during infection, and by subverting the normal stepwise generation of membranes during infection, SAI shows that with the right context and timing, induction of autophagy can be a negative for picornaviruses.
Results
SAI restricts EV-D68 infection
Several picornaviruses, including EV-D68, are known to hijack the cellular autophagy machinery to benefit multiple steps of their life cycle, including RNA replication, nonlytic release, and capsid maturation. However, the interplay between these viruses and autophagy is complex. While several members of the Piconarviridae family induce the formation of the double-membrane autophagosomes, they also block the fusion of autophagosomes and lysosomes, preventing cargo degradation. Indeed, we and others have recently shown that EV-D68 employs several strategies to obstruct autophagic flux [Citation15]. For this reason, we hypothesized that inducing flux immediately upon initiation of EV-D68 infection might attenuate viral replication. To this end, we treated H1HeLa cells with starvation media before (SBI) or after (SAI) viral infection, followed by viral titer determination by plaque assay. While SBI did not significantly impact the viral titer compared to the infection-only control, SAI reduced both the intracellular () and extracellular () viral titers and protected cells from EV-D68-mediated cytopathic effects ().
Figure 1. Starvation after infection restricts EV-D68 infection. H1HeLa cells were infected with EV-D68 (Infection Only), pretreated with starvation media for 4 h before infection, then fed (SBI), or treated with starvation media at 1 h post adsorption (SAI) for 4 h. The intracellular (a) and extracellular (b) viral particles were collected at 5 h post-infection for viral titer determination by plaque assay. (c) H1HeLa cells were infected with EV-D68 (infection only) or infected and treated at 0 hpi with either resveratrol (RES) or carbonyl cyanide m-chlorophenylhydrazone (CCCP) for 4 h. The intracellular titers were determined by a plaque assay. (d) H1HeLa cells were infected with EV-D68 (infection only), starved before infection (SBI), or starved at 0 h, 1 h, and 2 h post infection followed by intracellular viral titer determination by a plaque assay. (e) Cells were infected for the indicated time points in the presence of DMEM or starvation media before being subjected to a plaque assay. (f) H1HeLa cells were left uninfected (mock), treated with the starvation media (Starvation only), infected with EV-D68 (infection), or starved before (SBI) or after (SAI) infection. Images were captured using an EVOS microscope. Bars: 12.5 µm. An MOI of 30 was used for all infections. Error bars indicate mean ± SEM. n = 3 independent repeats for all experiments. Unpaired student’s t-test was used for the statistical analyzes (*** = p < 0.001; ** = p < 0.01;* = p ≤ 0.05; ns = not significant.).
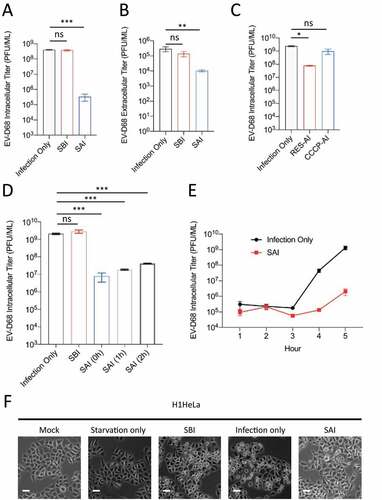
To determine how long post-infection this effect would still be observed, cells were infected, and starvation media was added 0, 1, or 2 hours post-infection (hpi). At all timepoints, SAI significantly decreased EV-D68 intracellular titers (). To further substantiate the effect of SAI on EV-D68 replication, we starved the cells immediately post adsorption, then titered cell-associated virus at one to five hours post-infection. This growth curve in shows that production of EV-D68 was delayed during starvation.
To verify that the effect of SAI on EV-D68 replication is not specific to starvation-induced autophagy, we treated the cells post-infection with resveratrol (RES) (a known autophagy inducer) and CCCP (a chemical that blocks autophagic flux) after infection for a plaque assay. As shown in , RES, not CCCP, diminished EV-D68 replication. Together, these results show that in contrast to SBI, which did not significantly alter EV-D68 replication, SAI potently decreased EV-D68 reproduction.
SAI overrides EV-D68-mediated blockage of autophagic flux
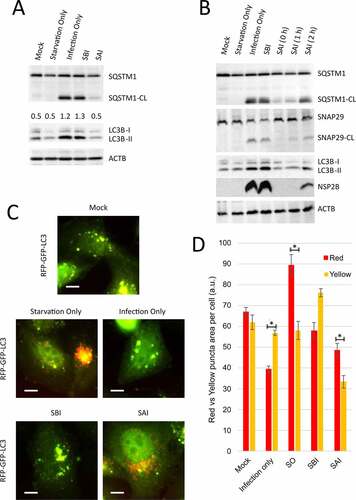
We have previously demonstrated that EV-D68 cleaves SQSTM1/p62 (sequestosome 1) and SNAP29 at later time points of infection, which coincides with the accumulation of lipidated MAP1LC3B/LC3B, known as LC3B-II. This suggests that cleavage of SNAP29 and SQSTM1 inhibits autophagic flux during EV-D68 infection. Therefore, to understand how SAI disrupts EV-D68 infection, we examined SAI’s effect on the viral-mediated cleavage of the aforementioned proteins. H1HeLa cells were left untreated (mock), starved (starvation only), infected (infection only), or starved before and after infection for a western blot assay. Consistent with our previous findings, EV-D68 induced cleavage of both SQSTM1 and SNAP29 and caused the accumulation of LC3B-II. SBI did not significantly affect these phenomena (). In contrast, SAI abrogated the viral-mediated cleavage of these autophagic flux-related proteins and blocked nonstructural protein 2B (NSP2B) translation (); delaying starvation induction led to a partial rescue of both SQSTM1 and SNAP29 cleavages and NSP2B expression (). Interestingly, we observed that SAI, similar to starvation only control, decreased LC3B-II expression to the basal level, indicating that SAI induced autophagic flux during EV-D68 infection (). To confirm that our SAI protocol induces autophagic flux during EV-D68 infection, we transfected the cells with an RFP-EGFP-LC3B construct and treated/infected them as in . While GFP signal is quenched in autolysosomes due to its acid lability, RFP signal is acid-resistant. Hence colocalization of the green and red signals into yellow puncta denotes autophagosomes, and singular red puncta represent amphisomes and autolysosomes. As indicated in , EV-D68 infection triggered the accumulation of yellow puncta, which was not substantially affected by SBI. In contrast, starvation and SAI both induced the accumulation of red puncta. These changes are quantified in by measuring the total area of red puncta vs. red+green (yellow) puncta.
Figure 2. SAI induces autophagic flux during EV-D68 infection. (a) Cells were treated/infected as described in . Lysates were collected at 5 hpi and analyzed by western blot against the indicated proteins. (b) Cells were mock-infected or infected then starved for 2 h pre-infection (SBI), or starved post-infection and fed with normal media at the indicated time point (SAI, 0, 1, or 2 h) and subsequently subjected to western blot analyses. (c) Cells were transfected with the RFP-GFP-LC3B plasmid overnight and left uninfected (mock), treated with the starvation media (Starvation only), infected with EV-D68 at an MOI of 30 (infection), or starved before (SBI) or after (SAI) infection. The cells were fixed at the end of the infection and examined by fluorescence microscopy. Bars: 3.5 µm. (d) Quantification of area of red (acidic) and yellow (nonacidic) puncta. (* = p value < 0.05; n = 3.) An MOI of 30 for 4 h was used for all infections.
SAI degrades autophagosomes and attenuates EV-D68 RNA replication
Many viruses are known to use complex membrane vesicular structures for RNA replication, often derived from the autophagy machinery [Citation17–20]. Since SAI induces autophagic flux during EV-D68 replication, we asked whether our SAI protocol prevents the accumulation of autophagosomes, thereby depriving the virus key replication organelles. To address this question, we starved H1HeLa cells before or after infection for electron microscopy analysis. As depicted in , mock infection did not induce the formation of autophagosome-like structures, but EV-D68 infection led to the accumulation of vesicles with the approximate size and electron-light interiors of later-infection autophagy-derived vesicles, as opposed to the tubular, electron-dense structures seen as early-infection replication organelles for picornaviruses [Citation6,Citation8]. The physical appearance of these structures was not impacted by our SBI protocol. In contrast, we do not observe accumulation of autophagosomes during our SAI protocol. To gauge the differential kinetics of autophagosome formation and accumulation due to SAI, we infected H1HeLa cells with and without starvation for western analysis against LC3B and SQSTM1. As shown in , EV-D68 infection triggered LC3B-II accumulation beginning at 3 h post-infection.
Figure 3. SAI degrades autophagosomes and inhibits viral RNA replication. (a) Cells were mock- infected, infected with EV-D68 (Infection only) for 4 h, or starved before (SBI) and after (SAI) infection. The cells were then fixed and analyzed by EM. Scale bar: 500 nm. (b) H1HeLa cells were infected for the indicated time points in the presence and absence of starvation for western blot (c) Cells were infected, then normal media or starvation media were added. Cells were then collected at the indicated time points, and total RNA was extracted and subjected to qPCR RNA analysis following cDNA synthesis. (d and e) H1HeLa cells were transfected with the indicated siRNA or the scramble control for 48 h, before being subjected to western blot analyses against the indicated proteins. (f and g) Cells were infected and cell-associated titers obtained at 5 hpi. For all experiments, MOI = 30, except for F and G, for which MOI = 0.1. Unpaired student’s t-test was used for the statistical analyzes. (*** = p < 0.001; ** = p < 0.01; * = p ≤ 0.05; ns = not significant.).
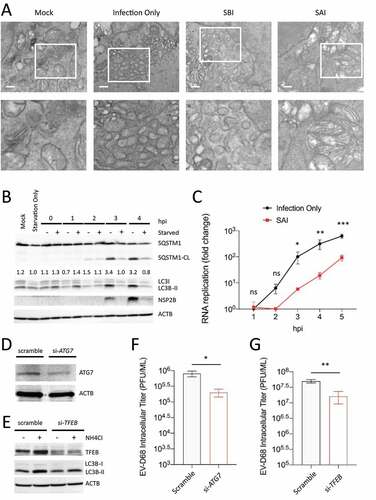
In contrast, induction of LC3B accumulation under our SAI protocol was quick and transient: it triggers LC3B-II accumulation at 1 h post-infection, which was cleared by 2 h post-infection, suggesting that SAI induces complete autophagic flux within 2 h post-infection. Given the importance of autophagosomes for picornavirus RNA replication and because SAI prevents the accumulation of these vesicles, we reasoned that SAI might attenuate viral RNA replication. To confirm this hypothesis, we infected H1HeLa cells with and without starvation media for various time points for RNA isolation and cDNA synthesis. Our qPCR results indicate that SAI robustly reduced EV-D68 replication (). To further understand the significance of this transient induction of autophagic signals for EV-D68 reproduction, we generated a knockdown (KD) of TFEB (transcription factor EB), the master transcriptional regulator of autophagy and lysosomal biogenesis, and measured the viral titer following low MOI infection [Citation21]. Our results show that KD of TFEB impeded the autophagic signaling and reduced EV-D68 replication (). Knockdown of ATG7 () also decreased viral titer (). Together, these results suggest that SAI attenuates EV-D68 replication by inducing a rapid autophagic response that prevents the formation of autophagosomes essential later in the viral life cycle.
SAI restricts EV-D68 replication in physiologically relevant cell lines
To ensure that the effect of our SAI protocol on EV-D68 replication and proteolytic activity is not cell type-dependent, we infected A549 lung cancer cells and differentiated SH-SY5Y neuronal cells with EV-D68, followed by a western blot analysis. As shown in , EV-D68 infection induces SQSTM1 cleavage and decreased SNAP29 expression in A549 cells. While SBI appears to enhance 2B expression, SQSTM1 cleavage, and loss of full-length SNAP29, SAI abrogated expression of 2B and SQSTM1 cleavage while restoring SNAP29 expression to basal levels. Similar results were observed in the differentiated SH-SY5Y cells, wherein SAI abrogated SQSTM1 cleavage and restored SNAP29 expression (). Interestingly, SBI appeared to attenuate the viral-mediated cleavage of SQSTM1 and the induction of LC3B-II accumulation in the SH-SY5Y cells (), suggesting that these cells are more sensitive to starvation and recover slower relative to H1HeLa and A549 cells.
Figure 4. SAI blocks EV-D68 replication in A549 and differentiated SH-SY5Y cells. (a) A549 cells, and (b) differentiated SH-SY5Y cells were treated/infected as in for western blot analyzes against the indicated proteins. (c) A549 cells were either infected with the virus only (Infection Only), pretreated with the starvation media before infection, or treated with the starvation media after viral adsorption until the end of the incubation. Intracellular viral particles were then collected and subjected to a plaque assay. (d) Differentiated SH-SY5Y cells were infected as in C, followed by a plaque assay. Error bars indicate mean ± SEM. Statistical analysis was done using unpaired student’s t-test. (** = p < 0.01; ns = not significant.).
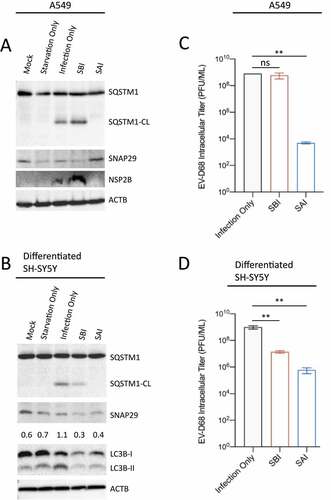
Furthermore, analysis of the viral titer in both A549 and differentiated SH-SY5Y cells reveals that SAI disrupted EV-D68 replication in these cells (). In contrast to the A549 cells in which SBI did not impact the viral titer, SBI significantly decreased EV-D68 replication in the differentiated SH-SY5Y cells, which could partly explain the SBI-mediated attenuation of both SQSTM1 cleavage and LC3B-II observed in . Together, these results suggest that SAI’s effect on EV-D68 replication is not cell type-dependent.
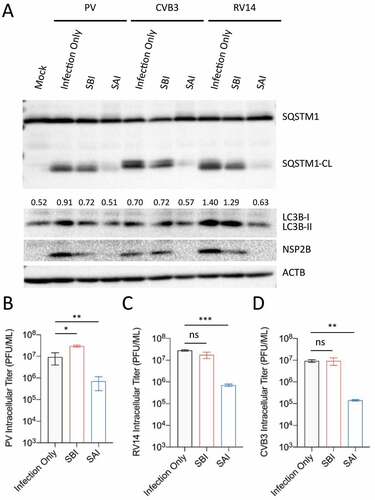
SAI impedes the replication of several medically important picornaviruses
Given the significance of autophagy for picornavirus replication, we asked whether SAI could restrict the replication of medically relevant picornaviruses, such as poliovirus (PV), coxsackievirus (CVB3), and rhinovirus 14 (RV14). We first examined whether SAI could abrogate SQSTM1 cleavage induced by these viruses. For this purpose, H1HeLa cells were starved before or after infection for western blot analysis. Similar to EV-D68, these viruses induce LC3B accumulation and cleave SQSTM1 (). In contrast to SBI, which did not significantly affect these phenomena, SAI abrogated SQSTM1 cleavage and attenuated LC3B-II accumulation for all viruses (). Finally, we examined the effect of SAI on the replication of these viruses. Our plaque assay data showed that SAI inhibited the replication of PV, CVB3, and RV14 to a similar extent, indicating that all these viruses require functional autophagosomes for their replication. In contrast, SBI did not significantly alter the titer of these viruses, except for PV, for which SBI increases viral replication ().
Figure 5. SAI attenuates the replication of several picornaviruses. (a) H1HeLa cells were mock infected or infected (MOI = 30 for 4 h) then fed or starved as indicated for western blot analysis. (b, c, and d) Cells were starved before (4 h) or after (5 h) infection with an MOI of 0.1 for 5 h. Lysates were then analyzed by plaque assay. Error bars indicate mean ± SEM and unpaired student’s t-test was used for statistical analyzes. (** = p < 0.01; * = p ≤ 0.05; ns = not significant.).
Neither blocking autophagic flux, nor amino acid supplementation, rescued viral replication
Although autophagy is mostly proviral during picornavirus replication, recent evidence suggests that the cellular process could target specific viral proteins for degradation through virophagy, thereby decreasing the viral titer [Citation22–24]. Because SAI induces autophagic flux during EV-D68 replication, we asked whether autophagy could degrade viral components. We reasoned that if SAI degrades viral components (viral RNA or protein), blocking SAI-induced flux should rescue the viral titer. For this purpose, we treated the cells after infection with and without the starvation media in the presence and absence of chloroquine for western blot analyzes. Chloroquine (CQ) is a lysosomotropic agent that increases lysosomal pH and hence blocks autophagic degradation. The result in showed that the addition of CQ in both fed and starved conditions stopped autophagic flux, as demonstrated by the buildup of SQSTM1 and LC3B-II.
Figure 6. Blocking autophagic flux or supplementing amino acids does not rescue SAI inhibition of EV-D68 infection. (a) H1HeLa cells plated in 6-well plates were infected/treated as indicated. The whole-cell lysates were then analyzed by western blot against the indicated proteins. (b) Cells were left untreated or treated with 100 μM CQ before and after infection. The intracellular viral particles were collected following three freeze-thaw cycles for a plaque assay. (c) Uninfected cells were treated with BAF alone for 4 h, or 100 nM BAF in starvation media for 4 h. Lysates were then analyzed by western blot. (d) H1HeLa cells were infected and starved-after-infection in the presence and absence of BAF. Cells were collected at the end of the infection for viral titer determination by a plaque assay. (e) Uninfected cells were starved for 4 h with or without 0.1X essential and nonessential amino acids, followed by western blot analysis. (f) Cells were either infected, starved after infection, or starved after infection in the presence of 0.1X essential and nonessential amino acids. The viral titers were determined by a plaque assay. An MOI of 30 was used for A and B, while an MOI of 0.1 was used for D and F. 100 μM Chloroquine pretreatment = CQ (+). Chloroquine pretreatment and post treatment = CQ (++). Error bars indicate mean ± SEM and unpaired student’s t-test was used for statistical analyzes. (* = p ≤ 0.05; ns = not significant.).
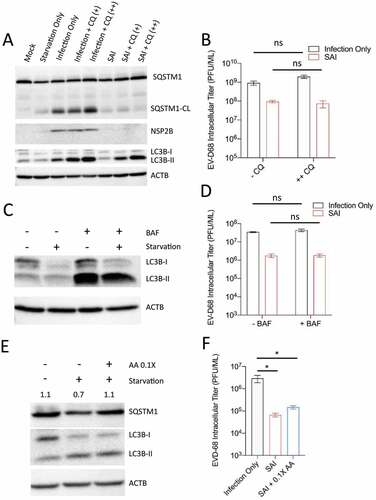
Interestingly, despite blocking autophagic flux during SAI, CQ treatment failed to rescue SQSTM1 cleavage (). We then examined the effect of CQ treatment on viral titer. Whereas CQ treatment marginally increased viral titer in the infection-only group, the drug treatment failed to rescue viral titer upon SAI (). We used bafilomycin A1 (BAF), a widely used vacuolar-type H+-ATPase inhibitor that stops autophagic flux. As expected, BAF treatment triggered LC3B-II accumulation in uninfected cells. () However, this same treatment did not rescue viral titer during SAI (). These results suggest that SAI does not target viral components for autophagic degradation.
Next, given that starvation media is an amino acid-deficient media and amino acids are required for translation, we reasoned that starving the cells after infection may attenuate viral translation, and decrease the overall viral titer [Citation25]. Our western blot results revealed that supplementing the starvation media with 10% of normal levels of essential and nonessential amino acids actually inhibited autophagic flux, as evidenced by an increase in SQSTM1 levels compared to regular starvation media (). To examine the effect of this amino acid supplementation on viral titer, we infected H1HeLa cells (MOI = 0.1) and starved them after infection with and without amino acid supplementation. As indicated in , amino acid supplementation did not rescue viral replication despite inhibiting flux, suggesting that the anti-EV-D68 activity of our SAI protocol does not implicate amino acid-dependent translation inhibition.
Discussion
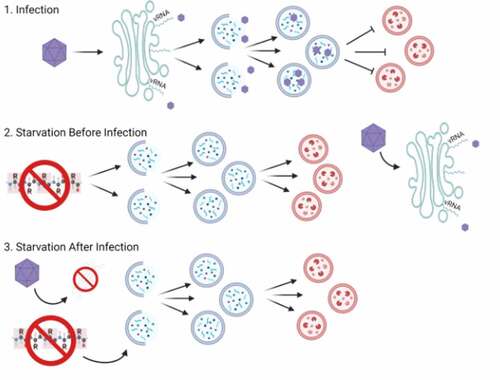
Our experiments demonstrate that inducing autophagosome formation immediately post-infection inhibits EV-D68 infection. This is somewhat counter-intuitive, since our lab and many other groups have shown that the autophagic machinery is required for efficient replication of many picornaviruses [Citation26–28]. However, we show that here as well: the ATG7 gene, required for efficient lipidation of the autophagosome resident protein LC3, is required for efficient replication of EV-D68 (.) We propose that the solution to this apparent paradox is in the limited pool of membrane-shaping resources in the cell. In our model, the cell activates autophagic stress pathways before the virus has a chance to access the autophagosome-making machinery. Our experiments suggest, as shown in our model in , that activating starvation-induced autophagy leaves cellular membrane-building reserves depleted.
Figure 7. Model of competition for membrane resources. 1. Infection. Picornaviruses induce complicated and unique membrane structures referred to as replication organelles. Double-membraned structures resembling autophagosomes are thought to bud from this organelle and provide harbors for virion maturation and egress from the cell. Viruses prevent these vesicles from fusing with lysosomes. 2. Starvation before infection (SBI). When cells are starved prior to adding virus, then fed after virus is adsorbed to the cells, most picornaviruses are unaffected (PV being the exception.) We propose that for most viruses, after a complete autophagic response, the resources needed for generating virus replication membranes can be renewed quickly, and these viruses generate replication membranes as usual. 3. Starvation after infection (SAI). When infected cells are starved immediately after, or within a few hours of, viral adsorption, the cellular stress-induced autophagy response develops, diverting resources from the generation of viral replication organelles in favor of autophagosomes and attenuating virus RNA replication.
Our data show that, as for other picornaviruses, EV-D68 infection induces autophagosome formation. Starving cells before infection (SBI) did not significantly impact the virus-induced generation of autophagosomes. Starving cells after infection (SAI) prevented autophagosome accumulation and decreased viral replication (). To understand how SAI prevents autophagosome accumulation, we performed a time-course analysis. We observed that EV-D68 infection induces autophagosome formation at 3 hpi, which coincided with NSP2B expression, suggesting that viral RNA replication is required for autophagosome formation (). In contrast, autophagosomes could be detected as early as 1 hpi in our SAI protocol; however, by 2 hpi, the autophagosomes had been cleared from the cell (). One possibility is that virus replication components could be sequestered inside autophagosomes and unavailable for producing viral RNA and capsids. However, given that SAI prevented autophagosome accumulation, we think this is unlikely. Since SAI inhibited accumulation of autophagosomes prior to the peak of viral RNA replication, which typically occurs between 3–4 hpi for high MOI infection, we posit that SAI’s anti-EV-D68 activity involves attenuating viral RNA replication. Consistent with the above hypothesis, SAI decreased EV-D68 RNA replication ().
It is important to note the speed of the autophagic process. Studies have shown that the time between induction of autophagy and degradation is remarkably brief; as little as ten minutes in yeast, and thirty minutes in mammalian cells [Citation29,Citation30]. We propose that SAI induces a complete autophagic process from induction to degradation, before the virus has a chance to usurp the autophagy proteins, lipids, and membranes required to generate the convoluted membrane organelle for RNA replication. Several of the components required for these unique organelles have been identified, including phosphatidylinositol 4-phosphate and cholesterol, as well as two guanine nucleotide exchange factors for ARF1: ARFGEF1/BIG1-ARFGEF2/BIG2 and GBF1 [Citation31–33]. We hypothesize that limited stores of one or more of these are rate-limiting for building virus-specific membranes when cellular autophagy is induced (see model, .)
During infection with poliovirus, longitudinal studies of infection using electron tomography showed formation of a convoluted series of membranes at 3 hpi, with double membraned structures visible at 4 hpi and later [Citation6]. Tomographic reconstruction led to the conclusion that the double-membraned vesicles derive from the convoluted structures. We propose that SAI short-circuits this by generating double-membraned vesicles, using components of the autophagic machinery such as ATG7, depleting the resources for building replication membranes.
It was recently shown that a specific ATG7 mutation, ATG7A388T [Citation34], inhibits poliovirus-induced LC3 lipidation, although the effect of this mutation on cellular autophagy is unclear. The study also showed that ATG7 knockdown increased PV infection in a neuron-like cell line, so the cell-type specificity of picornavirus requirements for the LC3-lipidation machinery needs to be addressed further. Another recent publication suggested that the effect of autophagy on picornavirus replication is cell-type specific [Citation35]. However, in addition to restricting EV-D68 infection in H1HeLa cells, our SAI protocol similarly blocked EV-D68 replication in A549 lung cancer cells and differentiated SH-SY5Y cells, which display neuron-like morphology and express neuronal cell markers (Wagner et al, in preparation.) (). Given the association of EV-D68 infection with acute flaccid myelitis, our differentiated SH-SY5Y cells could help study how EV-D68 causes AFM. Indeed, we have shown that the differentiated SH-SY5Y cells are permissive to EV-D68 infection. Although there has been some controversy regarding whether EV-D68 Fermon can infect neuronal cells, these cells are readily infectable [Citation36,Citation37] and our SAI protocol obstructs EV-D68 infection in these cells. Interestingly, while only SAI obstructed EV-D68 replication in H1HeLa and A549 cells, both our SBI and SAI protocols robustly hampered EV-D68 replication in the differentiated SH-SY5Y cells (). These findings suggest that the differentiated SH-SY5Y cells are more sensitive to autophagy-inducing stimuli, possibly because they do not recover from autophagy as quickly. Modulating autophagy could constitute a plausible treatment strategy against EV-D68 paralysis disease.
One study mentioned above also demonstrated that the poliovirus capsid proteins contain an LC3-A like domain, which cannot be detected in sequence homology, but which drives the VP0 protein to autophagosomes through its VP4 domain [Citation35]. One model incorporating these data would be that inducing autophagy immediately upon infection draws nascent viral polypeptides into degradative autophagosomes, reducing their ability to induce infection. Autophagy plays an essential role in antiviral immunity by targeting viral components for degradation [Citation10]. An increasing body of evidence indicates that autophagy targets the structural proteins of several viruses for degradation. Recently, Wen et al. showed that the autophagy receptor SQSTM1 targets VP1 and VP3 of Seneca Valley virus, a picornavirus that causes vesicular disease and neonatal fatal loss in pigs, for degradation [Citation22]. While we did not examine the interaction between EV-D68 structural proteins and SQSTM1 during infection, blocking autophagic flux did not rescue EV-D68 titers upon SAI (). Our findings suggest that autophagy does not target viral components, including viral proteins, for degradation. Instead, the cellular process induces flux, preventing the virus from generating unique replication organelles and consequently decreasing viral replication.
In addition to impairing EV-D68 reproduction, our protocol also reduced the replication of essential picornaviruses, including PV, (), CVB3 (), and RV14 (). SAI also attenuated SQSTM1 cleavage and autophagosome marker (LC3B-II) accumulation during infection with these viruses (). These findings underscore the importance of the virus-induced stepwise development of autophagosomes for picornavirus replication. Notably, our SBI protocol did not substantially affect the viral titers for all viruses tested, but for PV, SBI significantly increased PV replication (). Consistent with this finding, inducing autophagy before PV infection was shown to increase PV titers [Citation7]. Exactly how inducing autophagy before infection enhances PV replication but not the other tested picornaviruses is unclear; it is possible that poliovirus is an outlier in terms of picornavirus relationships to autophagy.
In conclusion, our data show that cellular autophagy and virus-induced membrane formation compete with one another for cellular resources, and stress-induced autophagy post-infection can short-circuit virus replication. Future studies will focus on understanding the specific resources the virus requires to generate replication organelles.
Materials and methods
Cell culture and plasmids
H1HeLa (ATCC, CRL-1958) and A549 (ATCC,CCL-185) cells were cultured in DMEM (Gibco, 11,965–092) supplemented with 10% heat-inactivated fetal bovine saline (GemininBio, 100–106), 1x penicillin-streptomycin (Gibco, 10,378–016), and 1x sodium pyruvate (Gibco, 11,360–070) and incubated in a 5% CO2 incubator at 37°C. SH-SY5Y cells (ATCC, CRL-2266) were differentiated by gradually decreasing the percentage of heat-inactivated FBS and supplementing the cells with trans-retinoic acid (Fisher Scientific, AC207340010). After adapting to low FBS, the cells were grown at 37°C in a 5% CO2 in the final differentiation media, which contains neurobasal (Invitrogen, 21,103,049), 50x B-27 supplement (Invitrogen, 17,504,044), potassium chloride (KCI) supplement, glutamax-L (35,050,061), BDNF (brain derived neurotrophic factor; Cell Signaling Technologies, 3897), dibutyryl cAMP (SelleckChem, S7858), penicillin-streptomycin, and trans-retinoic acid. A manuscript is in preparation to provide detail and verification of this differentiation protocol. The RFP-GFP-LC3B tandem construct was obtained from Addgene (84,573; deposited by Noboru Mizushima) and was transfected into the cells using the Lipofectamine 2000 transfection reagent (Invitrogen, 11,668–019). The transfection complex was removed at 6 h post-transfection and replaced with basal media. Viral infection/treatment was initiated 24 h post DNA transfection.
Western blotting
Cells were grown in 6-well plates and lysed with RIPA buffer (Sigma, R0278) supplemented with cOmplete Tablets Mini Protease Inhibitor Cocktail (ROCHE, 11,836,170,001) at the end of the infection or treatment. Lysates were collected in microcentrifuge tubes and incubated on ice for at least 30 minutes before being clarified at 10,625 x g for 30 minutes. The lysates were then loaded to SDS-PAGE and transferred to polyvinylidene fluoride (PVDF; Bio-Rad,1,620,177) membranes. The membranes were blocked for 1 h with 5% skim milk, washed twice with TBST (100 mM Tris hydrochloride, pH 7.4, 2.5 M sodium chloride, and 0.125% Tween-20 [Sigma Aldrich, P1379]), and incubated with primary antibodies at a 1:1000 dilution overnight on a shaker at 4°C. The membranes were rewashed twice with TBST and incubated with the secondary antibody (1:2000) at room temperature for 1 h before being developed by western Lightning ECL. Images were acquired using ChemiDoc (Bio-Rad).
Antibodies
Mouse monoclonal anti-SQSTM1 antibody was purchased from Abnova (H00008878-M01). Rabbit polyclonal anti-LC3B (NB600-1384) and rabbit polyclonal anti-ACTB (NB600-532) antibodies were purchased from Novus. Rabbit monoclonal anti-SNAP29 antibody (ab138500) was bought from Abcam. Rabbit polyclonal anti-TFEB (4240), rabbit monoclonal anti-ATG7 (2631), and rabbit polyclonal enterovirus D68 antibodies (WJ3397769C) were bought from Cell Signaling Technology.
Viral infections
Viral infection was performed as previously described with some modifications. An MOI of 0.1 for 5 h (low) or 30 for 4 h (high) was used for all infections. Virus particles were diluted in serum-free media, added to the cells, and incubated at 37°C for 1 h. The cells were washed with PBS (Quality Biological, 114–058-101) twice and overlaid with basal media until the end of the infection. For starvation before infection, cells were washed in PBS then starved for 4 h with starvation media [Citation25]. This amino acid-deficient media is used to induce autophagic flux: 140 mM NaCl, 1 mM CaCl2, 1 mM MgCl2, 5 mM glucose, 20 mM HEPES, pH 7.4, and 1% BSA (Fisher bioreagents, CAS 9048–46-8). Starvation media was added before infection, after which they were infected as the infection-only group. For starvation after infection, the cells were infected for 1 h (adsorption) and washed twice with PBS before being overlaid with starvation media for 4 h. The cells were either fixed and prepared for immunofluorescence assay or electron microscopy or collected in microcentrifuge tubes and stored at −80°C for plaque assay-based viral titer determination.
Drug treatments
Bafilomycin A1 (BAF [Sigma, 88,899–55-2]) at 100 nM and chloroquine (CQ Sigma, 50–63-5) at 100 µM, in complete (DMEM) or starvation media were added to cells after adsorption until the end of the infection. The cells were subsequently collected for western blot, or plaque assay analyzes.
siRNA-mediated knockdowns
siRNAs were used to knockdown the ATG7 and TFEB autophagy-related genes. H1HeLa cells were grown to 40% confluency and transfected with the ATG7 siRNA (Sigma, 16,124,494), TFEB siRNA (ambion, 6616) or the scrambled siRNA (Mission siRNA universal negative control 1 [Sigma, SIC001]) using Lipofectamine 2000 and Opti-MEM (Gibco, 31,985–070). Briefly, for each well of a 6-well plate, 100 nM of the siRNAs and 5 µl of Lipofectamine (Invitrogen, 11,668–019) were separately incubated in Opti-MEM for 5 minutes, after which they were mixed and incubated for 20 min at room temperature. Then, the cells were PBS washed and incubated with the transfection mixture for 6 h, after which the transfection complex was replaced with complete media. Finally, the cells were processed for viral infection or knockdown efficiency determination by western blot at 48 h post transfections.
Plaque assays
For extracellular viral titer measurement, 1 ml of the supernatants were collected into Eppendorf tubes and stored at −80°C until use. For intracellular viral titers, cells were scraped using cell scrapers and transferred to Eppendorf tubes followed by three free-thaw cycles. The resulting lysates were serially diluted and added to H1HeLa for 30 minutes. The inoculums were then removed and overlaid with 2x MEM and 2% agar in a 1:1 ratio for 48 h.
RNA isolation and quantitative polymerase chain reaction (qPCR)
Total RNA was isolated from H1HeLa cells using TRIzol (Ambion, 15,596,026) according to the manufacturer’s instructions. Thermo Scientific RevertAid H Minus First Strand cDNA Synthesis Kit (K1632) was used to synthesize the cDNA following the removal of the genomic DNA. qPCR was done using KiCqStart SYBR qPCR Ready Mix (Sigma, KCQS01) with the 7500 Fast Dx Real-Time PCR Instrument (Applied Biosystems) according to the manufacturer’s protocols. The following primers, 5′ TAACCCGTGTGTAGCTTGG-3′ and 5′ -ATTAGCCGCATTCAGGGGC-3′, which are specific to the 5′ untranslated region (UTR), were used to amplify EV-D68. The gene expression results were normalized to GAPDH and plotted as relative expression compared to the 1 h infection-only time point.
Statistical analysis
All experiments presented within have been conducted a minimum of three times. Statistical analysis was done using GraphPad Prism software (Version 7.03). Values represent mean ± standard error of the mean (SEM) of at least 3 independent repeats, unless otherwise indicated. Samples were analyzed Student t-test for comparison, and statistical significance was set at a p-value of < 0.05.
Acknowledgments
The authors thank Sohha Arrianejad for technical assistance and discussions. We thank Joseph Mauban and University of Maryland School of Medicine Center for Innovative Biomedical Resources, Confocal Microscopy Core – Baltimore, Maryland for help with Core resources. was created with BioRender. K.M. is supported by NIH/NIAID grant T32AI095190. This work was supported by NIH/NIAID grants R01AI141359 and R01AI104928 to W.T.J.
Disclosure statement
The authors declare no competing financial interests.
Additional information
Funding
References
- Eshaghi A, Duvvuri VR, Isabel S, et al. Global distribution and evolutionary history of enterovirus D68, with emphasis on the 2014 Outbreak in Ontario, Canada. Front Microbiol. 2017;8:257.
- Holm-Hansen CC, Midgley SE, Fischer TK. Global emergence of enterovirus D68: a Systematic review. Lancet Infect Dis. 2016;16(5):e64–e75.
- Lang M, Mirand A, Savy N, et al. Acute Flaccid paralysis following enterovirus D68 associated Pneumonia, France, 2014. Euro Surveill. 2014;19(44). DOI:10.2807/1560-7917.es2014.19.44.20952
- Schieble JH, Fox VL, Lennette EH. Others A probable new human picornavirus associated with respiratory disease. Am J Epidemiol. 1967;85(2):297–310.
- Esposito S, Bosis S, Niesters H, et al. Enterovirus D68 Infection. Viruses. 2015;7(11):6043–6050.
- Belov GA, Nair V, Hansen BT, et al. Complex dynamic development of poliovirus membranous replication complexes. J Virol. 2012;86(1):302–312.
- Jackson WT, Giddings TH Jr, Taylor MP, et al. Subversion of cellular autophagosomal machinery by RNA viruses. PLoS Biol. 2005;3(5):e156.
- Jackson WT. Poliovirus-induced changes in cellular membranes throughout infection. Current Opinion in Virology. 2014;9:67–73.
- Shi J, Luo H. Interplay between the cellular autophagy machinery and positive-stranded RNA viruses. Acta Biochim Biophys Sin. 2012;44(5):375–384.
- Ahmad L, Mostowy S, Sancho-Shimizu V. Autophagy-virus interplay: from cell biology to human disease. Front Cell Dev Biol. 2018;6:155.
- Mannack LV, Lane JD. The autophagosome: current understanding of formation and maturation. Rrbc. 2015;5:39–58.
- Klionsky DJ, Abdel-Aziz AK, Abdelfatah S, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (4th edition)1. Autophagy. 2021;17:1–382.
- Wong HH, Sanyal S. Manipulation of autophagy by (+) RNA Viruses. Semin Cell Dev Biol. 2020;101:3–11.
- Sun D, Wen X, Wang M, et al. Apoptosis and autophagy in picornavirus infection. Front Microbiol. 2019;10:2032.
- Corona AK, Saulsbery HM, Corona Velazquez AF, et al. Enteroviruses remodel autophagic trafficking through regulation of host snare proteins to promote virus replication and cell exit. Cell Rep. 2018;22(12):3304–3314.
- Mohamud Y, Shi J, Qu J, et al. Enteroviral infection inhibits autophagic flux via disruption of the SNARE complex to enhance viral replication. Cell Rep. 2018;22(12):3292–3303.
- Snijder EJ, Limpens RWAL, de Wilde AH, et al. A unifying structural and functional model of the coronavirus replication organelle: tracking down RNA synthesis. PLoS Biol. 2020;18(6):e3000715.
- Ulasli M, Verheije MH, de Haan CAM, et al. Qualitative and quantitative ultrastructural analysis of the membrane rearrangements induced by coronavirus. Cell Microbiol. 2010;12(6):844–861.
- Sharma M, Bhattacharyya S, Nain M, et al. Japanese encephalitis virus replication is negatively regulated by autophagy and occurs on LC3-I- and EDEM1-containing membranes. Autophagy. 2014;10(9):1637–1651.
- Oudshoorn D, van der Hoeven B, Limpens RWAL, et al. Antiviral innate immune response interferes with the formation of replication-associated membrane structures induced by a positive-strand RNA virus. MBio. 2016;7(6):e01991–16.
- Settembre C, Di Malta C, Polito VA, et al. TFEB links autophagy to lysosomal biogenesis. Science. 2011;332(6036):1429–1433.
- Wen W, Li X, Yin M, et al. Selective autophagy receptor SQSTM1/ p62 inhibits seneca valley virus replication by targeting viral VP1 and VP3. Autophagy. 2021 17 ;1–13.
- Judith D, Mostowy S, Bourai M, et al. Species-specific impact of the autophagy machinery on chikungunya virus infection. EMBO Rep. 2013;14(6):534–544.
- Orvedahl A, MacPherson S, Sumpter R Jr, et al. Autophagy protects against sindbis virus infection of the central nervous system. Cell Host Microbe. 2010;7(2):115–127.
- Axe EL, Walker SA, Manifava M, et al. Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-Phosphate and dynamically connected to the endoplasmic reticulum. J Cell Biol. 2008;182(4):685–701.
- Berryman S, Brooks E, Burman A, et al. Foot-and-mouth disease virus induces autophagosomes during cell entry via a class iii phosphatidylinositol 3-kinase-independent pathway. J Virol. 2012;86(23):12940–12953.
- Mauthe M, Langereis M, Jung J, et al. An siRNA screen for ATG protein depletion reveals the extent of the unconventional functions of the autophagy proteome in virus replication. J Cell Biol. 2016;214(5):619–635.
- Alirezaei M, Flynn CT, Whitton JL. Interactions between enteroviruses and autophagy in vivo. Autophagy. 2012;8(6):973–975.
- Xie Z, Nair U, Klionsky DJ. Atg8 controls phagophore expansion during autophagosome formation. Mol Biol Cell. 2008;19(8):3290–3298.
- Mejlvang J, Olsvik H, Svenning S, et al. Starvation induces rapid degradation of selective autophagy receptors by endosomal microautophagy. J Cell Biol. 2018;217(10):3640–3655.
- Belov GA, Altan-Bonnet N, Kovtunovych G, et al. Hijacking components of the cellular secretory pathway for replication of poliovirus RNA. J Virol. 2007;81(2):558–567.
- Richards AL, Soares-Martins JAP, Riddell GT, et al. Generation of unique poliovirus RNA replication organelles. MBio. 2014;5(2):e00833–13.
- Altan-Bonnet N. Lipid tales of viral replication and transmission. Trends Cell Biol. 2017;27(3):201–213.
- Brinck Andersen N-S, Jørgensen SE, Skipper KA, et al. Essential role of autophagy in restricting poliovirus infection revealed by identification of an ATG7 defect in a poliomyelitis patient. Autophagy. 2021;17(9):2449–2464.
- Zimina A, Viktorova EG, Moghimi S, et al. Interaction of poliovirus capsid proteins with the cellular autophagy pathway. Viruses. 2021;13(8):1587.
- Brown DM, Hixon AM, Oldfield LM, et al. Contemporary circulating enterovirus D68 strains have acquired the capacity for viral entry and replication in human neuronal cells. MBio. 2018;9(5). DOI:10.1128/mBio.01954-18
- Rosenfeld AB, Warren AL, Racaniello VR. Neurotropism of enterovirus D68 isolates is independent of Sialic Acid and is not a recently acquired phenotype. MBio. 2019;10(5):e02370–19.