
ABSTRACT
Macroautophagy/autophagy is a self-degradative process necessary for cells to maintain their energy balance during development and in response to nutrient deprivation. Autophagic processes are tightly regulated and have been found to be dysfunctional in several pathologies. Increasing experimental evidence points to the existence of an interplay between autophagy and cilia. Cilia are microtubule-based organelles protruding from the cell surface of mammalian cells that perform a variety of motile and sensory functions and, when dysfunctional, result in disorders known as ciliopathies. Indeed, selective autophagic degradation of ciliary proteins has been shown to control ciliogenesis and, conversely, cilia have been reported to control autophagy. Moreover, a growing number of players such as lysosomal and mitochondrial proteins are emerging as actors of the cilia-autophagy interplay. However, some of the published data on the cilia-autophagy axis are contradictory and indicate that we are just starting to understand the underlying molecular mechanisms. In this review, the current knowledge about this axis and challenges are discussed, as well as the implication for ciliopathies and autophagy-associated disorders.
Autophagy: the basics
Macroautophagy (hereafter referred as autophagy) is an evolutionarily conserved process used by cells to sequester intracellular components inside double-membrane organelles, called autophagosomes. These organelles fuse with lysosomes to degrade their content and recycle nutrients back to the cytoplasm (). Autophagy is a tightly regulated pathway that maintains cellular homeostasis, and it is crucial for defense against extracellular agents (e.g., pathogens)[Citation1]. Underscoring its essential role, autophagy dysfunction has been linked to a variety of human diseases (e.g., cancer, degenerative and immune disorders), as well as aging [Citation2–4].
Figure 1. Schematic representation of the autophagy process. (a) The phagophore expands while engulfing intracellular components, such as mitochondria, viral particles, protein aggregates, endoplasmic reticulum, to be degraded. The phagophore then closes to become an autophagosome characterized by a typical double membrane. The autophagosome later fuses with the lysosome to form an autolysosome that degrades the sequestered cytoplasmic material. (b) Selective cargoes are recognized by the autophagic receptors which are characterized by the ability to simultaneously interact with LC3/GABARAP and selective cargoes, thus bridging the growing phagophore and the targeted cargoes. Cargoes can be ubiquitinated. Autophagy receptors bind the ubiquitinated cargoes through ubiquitin-binding domains (UBD) (blue) and bind LC3 and/or GABARAP subfamily proteins through the LIR domain (LIR, Orange). Ub: ubiquitin.
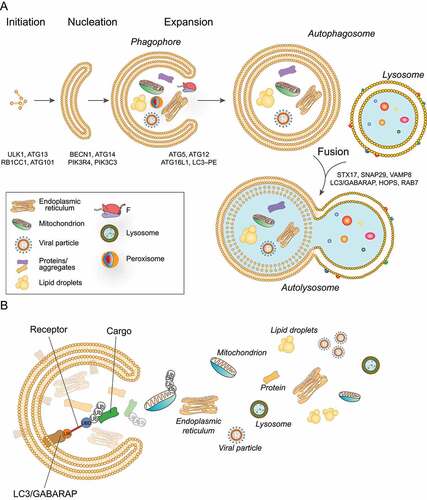
Autophagosomes are trafficked via microtubules and fuse with lysosomes, generating a single membrane organelle, the autolysosome [Citation5] (). Lysosomes, the destination of cargoes processed by autophagy, have an acidic pH and contain hydrolases with the capacity to degrade macromolecules, including lipids, proteins and sugars. The acidic pH is maintained by a proton pump, the multicomplex vacuolar-type H+-translocating ATPase (V-ATPase) [Citation6]. Upon fusion of autophagosomes with lysosomes, cargoes are degraded, and the resultant building blocks are subsequently released for anabolic reactions.
Autophagosome biogenesis is a process orchestrated by autophagy-related (ATG) proteins, and involves several steps including phagophore nucleation, expansion, and closure to allow sequestration of cytoplasmic cargoes [Citation7]. A number of upstream signaling complexes regulate autophagy, including MTOR (mechanistic target of rapamycin kinase) complex 1 (MTORC1), a serine-threonine kinase that positively regulates cell growth and proliferation by promoting anabolic activities, and by limiting catabolic processes such as autophagy [Citation8]. Under nutrient rich conditions, MTORC1 activity prevents autophagy by inhibition of the mammalian autophagy-initiating kinase ULK1 through phosphorylation; conversely, in instances of nutrient scarcity, ULK1 is activated by AMP activated protein kinase (AMPK) and promotes autophagy [Citation9]. Briefly, upon autophagy initiation, the ULK1 kinase complex translocates to the phagophore and recruits the class III phosphatidylinositol 3-kinase (PtdIns3K) complex which produces the signaling lipid phosphatidylinositol-3-phosphate (PtdIns3P) in the phagophore membrane. The signal generated coordinates the recruitment and activation of the autophagic cascade machinery to orchestrate autophagosome formation [Citation7].
Importantly, although autophagy was initially viewed as primarily a nonselective intracellular degradation pathway, recent studies have shown that autophagy selectively degrades substrates. Indeed, selective autophagy is required to maintaining the number and integrity of cellular organelles, as well as to protect cells from pathogens. This process consists of the specific targeting of cargoes for degradation, including proteins and unwanted/damaged organelles such as mitochondria, portions of the ER, ribosomes, peroxisomes, lysosomes among others [Citation10] (). Most cargoes are recognized by the Atg8-family proteins. These are core autophagic proteins associated with the autophagosome membranes via covalent conjugation to phosphatidylethanolamine. Family members include the MAP1LC3 (microtubule associated protein 1 light chain 3; LC3A, LC3B, LC3B2, LC3C) and the GABARAP (GABA type A receptor-associated protein; GABARAP, GABARAPL1, and GABARAPL2) sub-families (). Atg8 proteins are characterized by 15- to 20-amino-acid-long sequence motifs, termed LC3-interacting regions (LIRs) [Citation10]. In some cases, the cargo is recognized by selective autophagy receptors, which are characterized by the ability to bind cargoes and facilitate the recruitment of autophagic machinery and the formation of the growing phagophore [Citation11] (). Autophagy receptors bind Atg8-family proteins through a LIR domain and are finally degraded with the cargo. In mammals, over 30 autophagy receptors, such as SQSTM1/p62 (sequestosome 1), NBR1 (NBR1 autophagy cargo receptor), CALCOCO2 (calcium binding and coiled-coil domain 2), OPTN (optineurin) and TAX1BP1 (Tax1 binding protein 1) have been described. Their action is regulated by protein modifications, predominantly phosphorylation, ubiquitination, acetylation and oligomerization in order to operate in a specific type of autophagy, and/or to facilitate binding to other proteins [Citation10,Citation12]. Furthermore, selective autophagy can be classified as ubiquitination-dependent or -independent. In the first, cargoes are ubiquitinated: the ubiquitin is usually exposed to the cytosol and is captured by the receptors through a ubiquitin-binding domain. Receptors in turn bind LC3/GABARAP proteins through the LIR [Citation11] ().
Cilia biology
Cilia are microtubule-based organelles protruding from the cell surface and come in two flavors: motile and immotile. Motile cilia are present in single or multiple copies (> 100) per cell, beating in coordinated waves to propel liquids and/or mucus across the tissue surface[Citation13–15]. Motile cilia display different functions depending on the organ context and according to the cell type. One example is the respiratory epithelium where cilia on epithelial cells constantly move mucus up from the lungs to the back of the throat. Another example is given by the fallopian tubes, where cilia move the eggs toward the uterus. In the embryonic node, also known as the left-right organizer in vertebrates, motile cilia generate a directional fluid flow responsible for determination of the left-right (L-R) axis [Citation16]. Such flow has been well studied in animal models (mice, frogs and zebrafish) and has been found to promote asymmetric gene expression ultimately leading to asymmetric activation of specific developmental signaling pathways that lead to vertebrate visceral asymmetry [Citation16]. In addition to organ-specific motile cilia, most cells of the body also display a single immotile cilium known as the primary cilium with mechanosensory and chemosensory properties that allows the detection, integration, and transduction of a variety of extracellular signals. These signaling activities are carried out by transmembrane receptors embedded in the cilium membrane and by a myriad of signaling molecules localized to the primary cilium, which allow the cell to respond to various external stimuli and control the signaling pathways. The extracellular signals conveyed by primary cilia include mechanical stresses (e.g., bending of the cilium by fluid flow and tissue deformation), paracrine signaling molecules (e.g., HH [hedgehog], WNT, NOTCH, Hippo), growth factors and hormones. In specialized cells (e.g., kidney epithelial cells and photoreceptors), primary cilia can respond to specific stimuli, including force, light, temperature, gravity, and osmolarity. Moreover, regulation of primary ciliogenesis is coordinated with mitosis, implying the association of primary cilia with control of cell proliferation. Indeed, ciliogenesis is tightly correlated with the cell cycle, as the formation of a primary cilium occurs post-mitotically in G0 or early G1 phase, while its reabsorption occurs in S or G2 phase.
Both motile and immotile cilia have a microtubule-based structure formed by an axoneme composed of 9 microtubule doublets, anchored to the cell through a basal body (). Most motile cilia have typically a central pair of microtubules with a “9 + 2” structure (nodal cilia lack the central pair) associated with dynein arms that power axonemal beating through ATP hydrolysis. Primary cilia lack this central pair and dynein arms; they have a “9 + 0” structure and therefore lack motility ().
Figure 2. Cilia structure. Left: the ciliary axoneme is composed by nine microtubule doublets of microtubules (blue rods), which nucleate from the basal body (mother centriole). The axoneme protrudes from the cell membrane (ciliary membrane). The movement of IFT particles to the tip of the flagellum is powered by kinesin-II, a microtubule-based molecular motor. The movement of IFT particles back to the base of the flagellum is driven by the cytoplasmic dynein 2, another microtubule-based molecular motor. The mother and daughter centrioles are indicated by the blue cylinders and the transition zone by green at the bottom of the axoneme. The transition zone mediates interactions with the ciliary membrane. Right, top: motile cilia display a central microtubule pair connected with the outer microtubular doublets by radial spokes. The outer microtubules are connected by the nexin-dynein complex. Other structures include the axonemal inner and outer dynein arms. Right, bottom: Immotile primary cilia display nine peripheral microtubular doublets with no central pair and without dynein arms.
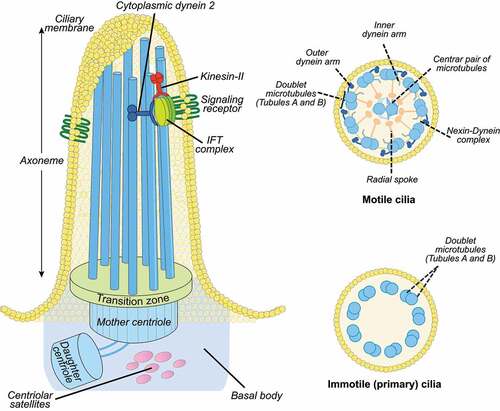
The basal body originates from the centrosome [Citation17], a cytoplasmic organelle composed of two centrioles (mother and daughter) and centriolar satellites, which are granular structures of pericentriolar material (PCM) (reviewed in [Citation18]) (). A specialized ciliary domain, the transition zone, is localized at the base of cilia and exerts a gate function for protein and lipid exit and entry, thus controlling the protein and lipid composition of the ciliary compartment [Citation19]. Cilia rely on intraflagellar transport (IFT), which uses kinesin and dynein motors for the supply of ciliary components synthesized in the cytoplasm and transported from the cell body to the ciliary tip and backwards [Citation20]().
For a detailed description of cilia structure, assembly, maintenance and recent advances in the understanding of the cell biology of this organelle, readers can refer to other excellent reviews [Citation21,Citation22]. Hundreds of proteins have been linked to ciliogenesis. Moreover, genes encoding some of these proteins have been associated with inherited conditions known as ciliopathies. Ciliopathies involving primary cilia often present multi-organ phenotypes that affect the central nervous system, eye and skeleton as well as visceral organs including the kidneys (primary ciliopathies) [Citation23]. These disorders include more common conditions such as autosomal dominant polycystic kidney disease (ADPKD) [Citation24]and rare disorders including oral-facial-digital type I (OFDI) [Citation25–27], Joubert (JBTS) [Citation28], Meckel-Grouber (MKS) [Citation29], Birt-Hogg-Dubé (BHD) syndromes [Citation30] and nephronophthisis (NPHP) [Citation23,Citation31]. Moreover, a large body of evidence has demonstrated that primary cilia are dysfunctional in human cancers, including melanoma, breast, pancreatic and renal cell cancers, cholangiocarcinoma and Hurthle cell carcinoma [Citation32–34]. Furthermore, dysfunction of motile cilia results in motile ciliopathies ranging from isolated laterality defects or subfertility in males, to primary ciliary dyskinesia (PCD), which is a multisystemic disorder that represents the most common motile ciliopathy with a global estimated prevalence of 1:10,000 [Citation35].
Autophagy and cilia come together
A tight link between primary cilia and autophagy was recently established. Indeed, autophagy has been shown to regulate ciliogenesis, and conversely, cilia-dependent signaling pathways and ciliary proteins are now known to control autophagy [Citation36–39].
Selective autophagy takes action in ciliogenesis
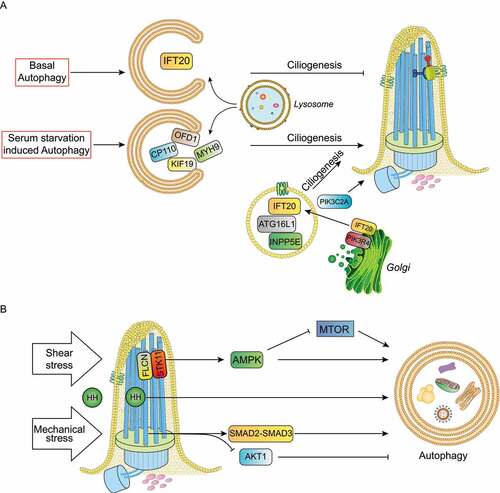
The first indication that autophagy controls ciliogenesis was provided in 2013 by Tang and colleagues, who demonstrated that serum starvation-induced autophagy promotes ciliogenesis through selective degradation of the satellite pool of the OFD1 protein, encoded by the gene responsible for OFD type I syndrome [Citation40]. OFD1 localizes to centrosomes/basal bodies, as well as centriolar satellites. In particular, it has been shown that the centrosomal/basal body pool of OFD1 is instead necessary for the formation of distal appendages of the mother centriole, a process required for IFT88 (Intraflagellar Transport 88 protein) recruitment, which in turn is necessary for cilia formation [Citation41]. Moreover, characterization of the ofd1 knockout mouse model, which recapitulates the human disorder, established a role for OFD1 in primary cilia assembly [Citation42]. In contrast to data reported by Tang et al., in the same issue, Pampliega and colleagues showed that basal autophagy inhibits ciliogenesis in mouse embryonic fibroblasts (MEFs) by the degradation of IFT20 (intraflagellar transport 20), an essential component of ciliary transport [Citation43]. To reconcile these observations, the authors proposed that basal autophagy limits ciliogenesis by removing IFT20, while autophagy induced by serum starvation enhances ciliogenesis by removing the satellite pool of OFD1().
Figure 3. Cilia-autophagy crosstalk models. (a) Under serum starvation selective autophagy degrades ciliary proteins such as OFD1, MYH9, CP110 and KIF19 to promote ciliogenesis and cilia elongation. Conversely, basal autophagy inhibits ciliogenesis by removing IFT20. ATG16L1, INPP5E and IFT20 trafficking to cilia affects primary cilia organization and signaling independently of their functions in autophagy. PIK3C2A and ATG16L1 control ciliogenesis via PtdIns3P. (b) HH (hedgehog) ligands activate HH signaling transduction and autophagy; shear stress in kidney epithelial cells activate the FLCN-STK11-AMPK-MTORC1 cascade and the mechanical stress activates the autophagy stimulatory SMAD2-SMAD3 pathway and the inhibitory AKT1 signaling in trabecular meshwork cells.
Following these initial observations, various laboratories demonstrated that autophagic degradation can control ciliogenesis [Citation32,Citation38,Citation44]. Another player in the selective autophagy-cilia association is the cytosolic HDAC6 (histone deacetylase 6). HDAC6 has been identified as an important cytoplasmic tubulin deacetylase that influences mitosis and chemotaxis through the regulation of tubulin stability [Citation45] and as an enhancer of autophagosome-lysosome fusion [Citation46]. Lam et al. reported that increased autophagy in cultured primary airway epithelial cells under chronic cigarette smoke exposure is associated with HDAC6-mediated motile cilia shortening and increased amounts of ciliary proteins (e.g., IFT88, ARL13, CETN1 [centrin 1] and PCNT [pericentrin]) localized to autophagosome fractions in the mouse airway. Indeed, the motile cilia phenotype caused by cigarette smoke exposure results in bronchitis associated with airway inflammation, mucus obstruction and emphysema [Citation47].
Three additional studies were recently published, further advancing the field. A decade ago it was shown that the removal of the CP110 (centriolar coiled-coil protein 110) from mother centrioles is required for ciliogenesis initiation after serum starvation [Citation48,Citation49]. However, the precise mechanism was unknown. Liu et al. have now identified NUDCD2/NudCL2 (NudC domain containing 2) as a novel selective autophagy receptor which mediates the removal of CP110 from mother centrioles through autophagy. The authors demonstrated that NudCL2 contains a LIR motif mediating the association of CP110 with LC3, and that this interaction occurs specifically upon serum deprivation to initiate ciliogenesis [Citation50].
The second very recent example of selective autophagy with a role in ciliogenesis is given by NEK9 (NIMA related kinase 9) which functions as a selective autophagy receptor for MYH9 (myosin heavy chain 9), a negative regulator of ciliogenesis. NEK9-mediated autophagic degradation of MYH9 induces ciliogenesis by promoting actin dynamics. Interestingly, Yamamoto and colleagues showed that NEK9 contains a LIR domain that, when mutated, impairs cilia formation in murine kidneys [Citation39].
In the third report, Arora et al., discovered that autophagy also modulates motile cilia growth, a highly regulated process important for flow generation [Citation51]. Cilia length is crucial for mucociliary clearance [Citation52,Citation53], in Chlamydomonas reinhardtii where the beating of cilia occurs only after a critical length is reached [Citation54], and in zebrafish, where altered cilia length results in perturbation of fluid flow generation [Citation55]. It was previously shown that KIF19/KIF19A (kinesin family member 19) residing at the ciliary tip, was essential for cilia length control [Citation56]. Arora and colleagues reported that ADCY6 (adenylate cyclase 6), known as a major supplier of cAMP in the modulation of primary cilia length [Citation57,Citation58], inhibits degradation of KIF19 by inhibiting autophagy [Citation51] (). Specifically, they showed that KIF19 interacts with AMPK, and that ADCY6 inhibits AMPK-KIF19 binding. In airway epithelial cells, in the absence of ADCY6, the activation of AMPK mobilizes KIF19 into autophagosomes for degradation. Indeed decreasing KIF19 levels correlate with elongated cilia and viceversa [Citation51]. Interestingly, experimental evidence suggests a length-dependent mechanical model for primary cilia. These findings add an additional layer of complexity in the autophagy-cilia interplay [Citation59].
Core autophagic proteins with an autophagy-independent role in ciliogenesis
The first example of a protein involved in autophagic processes with an autophagy-independent role in cilia comes from PIK3R4/VPS15, a regulatory subunit of the class III PtdIns 3-kinase PIK3C3/VPS34, belonging to the PtdIns3K complex [Citation60,Citation61]. Pampliega et al., showed that PIK3R4/VPS15 localizes to ciliary basal bodies and axonemes [Citation43]. Some years later, Stoetzel and colleagues demonstrated that, besides being required for autophagy, PIK3R4/VPS15 regulates primary cilium length in human fibroblasts, as well as ciliary processes in zebrafish [Citation62]. They showed that patients with mutations in PIK3R4/VPS15 display a ciliopathy phenotype characterized by skeletal abnormalities, retinitis pigmentosa and renal cysts. They also demonstrated that fibroblasts from patients have shorter cilia due to defective formation and/or release of IFT20-positive vesicles from the cis-Golgi to the primary cilium [Citation62].
Interestingly, ATG16L1 (autophagy related 16 like 1) which plays crucial roles in the conjugation of ATG8 protein, was also found to have non-autophagic functions. It was shown to interact with IFT20 both in nutrient-rich and serum deprivation conditions in ciliated and non-ciliated cells [Citation43,Citation63,Citation64]. Moreover, ATG16L1 was found to be co-transported with IFT20 to the cilium during ciliogenesis, and, just recently, the absence of ATG16L1 was shown to lead to the formation of giant cilia that are at least partially dysfunctional [Citation64]. The formation of this giant ciliary structures is caused by the absence of INPP5E (inositol polyphosphate-5-phosphatase E) in cilia with consequent accumulation of PtdIns(4,5)P2 instead of PtdIns4P . Thus, the authors demonstrated that ATG16L1 controls ciliary phosphoinositides (PIs) by regulating INPP5E-IFT20 trafficking to cilia, independently of its functions in autophagy, and affecting primary cilia organization and signaling functions [Citation64,Citation65].
Cilia influence autophagy
The first evidence that cilia act on autophagy was provided in 2013 by Ana Maria Cuervo<apos;>s group, who demonstrated that some ATG proteins display a ciliary or periciliary localization. In particular, cilia stain positive for proteins involved in the early steps of autophagosome formation (i.e., PIK3R4/VPS15, ATG16L1), components of the phagophore expansion complex (i.e., PIK3C3/VPS34) or markers of mature autophagosomes (i.e., LC3 and GABARAP), displaying a discrete punctate pattern at basal bodies and axonemes [Citation43]. Based on these results, it was proposed that the ciliary membrane represents a specialized site for autophagosome formation [Citation43]. However, BECN1 (beclin 1), a core component of the PtdIns3K complex, and ULK1, both key players in phagophore nucleation, do not localize to cilia, suggesting the existence of different autophagosome initiation complexes. Moreover, the same authors showed that autophagy activation upon serum starvation (which induces both ciliogenesis and autophagy), requires the presence of a functional primary cilium as Ift20- and Ift88-depleted cells (MEFs and kidney epithelial cells [KECs]) show defective ciliogenesis and decreased autophagy upon serum removal [Citation43]. In addition, it was shown that stimulation of cells with HH ligands concomitantly activates transcription of HH signaling target genes and autophagy [Citation43]. Autophagy overactivation was also observed in two additional experimental conditions in which HH signaling is constitutively active: in knockout cells for the negative regulator PTCH (patched), in which the HH signaling is constitutive activated, and in cells overexpressing GLI1, the main transcription factor for HH signaling target genes. Conversely, treatment with HH antagonists reduces autophagy [Citation43]. In addition, HH stimuli are unable to induce autophagy in cells with defective ciliogenesis, while GLI1 overexpression partially rescues autophagy loss in cilia-defective cells [Citation43]. After this initial report, a large body of experimental evidence has demonstrated that primary cilia-dependent autophagy can be activated by a variety of external stimuli (e.g., starvation, growth factors, mechanical stress) [Citation43,Citation66–70]. Jang and colleagues identified a primary cilium-autophagy-Nrf2 control axis that directs human embryonic stem cells (hESC) toward neuroectoderm (NE) fate. Indeed, they showed that increased ciliation in NE precursors induced autophagy resulting in inactivation of NFE2L2/Nrf2. Inhibition of NFE2L2, which directly binds to upstream regions of the pluripotency genes POU5F1 and NANOG to promote their expression, induces NE differentiation, thus demonstrating a key role for primary cilia-dependent autophagy in early steps of hESC differentiation [Citation71].
One of the first examples of mechanical stress acting on primary cilia and stimulating autophagy was described by Orhon and colleagues in 2016. In this first report, the authors demonstrated that in renal epithelial cells primary cilia, which transduce extracellular shear stress induced by fluid flow, ultimately induce STK11/LKB1 (serine/threonine kinase 11)-mediated AMPK activation and inactivate MTORC1. This stimulus results in autophagy upregulation and cell size reduction and, accordingly, cells with defective primary cilia show cellular hypertrophy [Citation67,Citation72]. A few years later, the same group demonstrated that FLCN (Folliculin), a component of the primary cilium, is upregulated upon shear stress and functions upstream of AMPK, orchestrating cell size reduction in KECs in an autophagy-dependent manner [Citation66]. Intriguingly, in KECs submitted to shear stress, primary cilium-dependent autophagy has been demonstrated to act independently of ULK1 and BECN1. PtdIns3P, the key lipid involved in autophagosome biogenesis, is produced by the basal body localized class II PIK3C2A [Citation73], which regulates ciliogenesis [Citation74] and promotes the synthesis of a local pool of PtdIns3P upon shear stress [Citation73]. Renal tubular epithelial cells from the Pik3c2a± mouse model show decreased autophagic activation and loss of cell-size control [Citation73]. However, how primary cilia control AMPK-mediated autophagy induction in the absence of phosphorylation of its targets – ULK1 and BECN1 – remains an open question. Another recent study showed the importance of primary cilia-mediated autophagy in physiological adaptation to stress in the context of pressure-induced autophagy in trabecular meshwork (TM) cells located in the anterior segment of the eye. Changes in pressure gradients and fluid flow associated with eye movement, circadian rhythm or the ocular pulse are translated in continuous cycles of tissue deformation and relaxation; TM cells can sense these deformations as mechanical forces and respond by reorganizing the actin cytoskeleton, modifying gene expression, secreting cytokines, and remodeling the extracellular-matrix (reviewed in [Citation75]). The activation of autophagy is one of the responses elicited in TM cells following application of mechanical stress [Citation76,Citation77]. Shim and colleagues reported that the primary cilium acts as a mechanosensor for mechanical stress-induced autophagy in TM cells, and identified AKT1 and SMAD2-SMAD3 as signaling pathways regulating the activation of this process which plays a critical role in intraocular pressure homeostasis [Citation76] (). A further demonstration of cilia acting on autophagy was provided by Xiang et al., in a study analyzing viability, proliferation and differentiation capacities of chondrocytes treated with cyclic tensile strain of different intensities. The authors found that mechanical stress promoted ciliogenesis and activated autophagy during cartilage development, and demonstrated that the integrity of primary cilia was of crucial importance for mechanical stress- regulated signal transduction and autophagy pathway [Citation68].
Core ciliary proteins with cilia-independent autophagy functions
A variety of proteins involved in ciliogenesis and mutated in ciliopathies, act directly in different stages of the autophagic cascade, independently of cilia. OFD1 represents a paradigmatic example of the level of complexity underlying the selective autophagy-cilia link. Indeed, it has been recently demonstrated that the OFD1 protein is not only degraded by autophagy to promote ciliogenesis, but also plays a direct role in the control of the autophagic cascade. Morleo and coauthors showed that OFD1 controls autophagosome biogenesis by promoting autophagic degradation of ATG13, a member of the ULK1 autophagy initiation complex, and that OFD1 acts as a novel autophagy receptor for ATG13 [Citation78]. The central portion of OFD1 specifically binds ATG13 while the LIR domain at the C terminus of the OFD1 protein mediates the direct interactions with ATG8 proteins [Citation78]. Furthermore, OFD1-mediated autophagic degradation of ATG13 inhibits autophagy independently of OFD1 cilia-related functions [Citation78]. However, it cannot be ruled out that OFD1 could also selectively recognize ciliary cargoes besides ATG13, and thus influence ciliogenesis through their selective degradation in a cell context-dependent fashion. This hypothesis is supported by recent evidence showing that different cilia components are selectively degraded through autophagy (see above)[Citation47]. Thus, OFD1 could be one of the players coordinating the intricate bidirectional crosstalk between autophagy and cilia, explaining the cellular need to compartmentalize autophagic and ciliary proteins to the same intracellular space [Citation38,Citation79]. Additionally PCM1 (pericentriolar material 1), which is necessary for ciliogenesis [Citation80,Citation81], is involved in GABARAP-autophagosome biogenesis independently of cilia [Citation82]. Joachim and colleagues showed that PCM1 physically interacts with GABARAP through a LIR motif and controls GABARAP localization and degradation at centriolar satellites thus influencing GABARAP-autophagosome formation [Citation82]. The experiments were performed in non-ciliated cells, leading the authors to hypothesize that also the role of GABARAP-PCM1 on autophagosome biogenesis is cilia-independent [Citation82].
Another ciliary protein that intervenes at early steps of autophagy in a cilia-independent manner is IFT20, an integral component of the IFT system. Finetti et al., showed that IFT20 regulates autophagy in non-ciliated T-cells, for which cell homeostasis, activation and differentiation relies on basal autophagy [Citation83]. The authors showed that IFT20 interacts with ATG16L1 and is required for the association of ATG16L1 with the Golgi complex and early endosomes, both of which have been identified as membrane sources for phagophore elongation. Moreover, they demonstrated that under basal conditions, IFT20 participates in autophagosome formation at the early endosome stage, by allowing recruitment of ATG16L1 and downstream autophagy regulators to early endosomes in T cells [Citation83].
Controversies in the cilia/autophagy interplay
The cilia-autophagy interplay has been controversial since its discovery [Citation40,Citation43] and contradictory findings demonstrating opposing roles for induced autophagy in ciliogenesis, have been described [Citation38]. Liu and coauthors reported that the transcription factors PPARA and NR1H4, which are nutrient-sensing receptors, induce ciliogenesis under fasting conditions or negatively-regulate cilia formation under nutrients, by controlling the expression of autophagy genes [Citation84]. PPARA activation facilitates autophagy by inducing expression of LC3, SESN2, ULK1, ATG2A, and ATG12 and by promoting ciliogenesis. In contrast, NR1H4 suppresses cilia formation by inhibition of autophagic gene expression [Citation84]. Chemical compounds such as sertraline and thioridazine, induce ciliogenesis through autophagy activation. In particular, treatment with sertraline or thioridazine increases the number of LC3 puncta in retinal pigmented epithelial (RPE) and cancer cells, and decreases SQSTM1 protein levels suggesting activation of the autophagic flux [Citation85,Citation86]. These findings are in line with those described by Tang in 2013. Conversely, Lam et al. suggested the term “ciliophagy” for the autophagy-mediated cilia disassembly through degradation of ciliary components such as IFT88 [Citation47]. Accordingly, Struchtrup showed that MEFs depleted for RPGRIP1L, a transition zone component, have reduced autophagic activity, which leads to cilia elongation [Citation87]. These findings were confirmed by treating cells with 3-methyladenine (3-MA), an inhibitor of early stages of autophagy, which leads to increased cilia length; conversely reduced cilia length was observed after treatment with the autophagy activators rapamycin or ABT-737 [Citation87].
Altogether, the abovementioned findings suggest that autophagic degradation could exert opposing effects on ciliogenesis in a context-dependent manner and that different players could be involved according to the cell type. However, contrasting findings have also been reported for experiments performed in the same cell line, hinting that culture conditions are important. An example is given by Wang, who showed that treatment with the autophagy inhibitors 3-MA, chloroquine or bafilomycin A1 (Baf) decreases cilia length, while treatment with autophagy inducers such as Tat-Beclin 1 and trehalose promotes ciliogenesis in human kidney 2 (HK2) cells [Citation88]. In contrast, Iaconis et al. demonstrated that treatment with Baf A1 and Tat-Beclin 1 exerts the opposite effect to those described by Wang et al. even though the experiments were performed in the same cell line [Citation89]. These observations suggest that autophagy may have a cell context-dependent role in cilia formation and maintenance. Indeed, autophagy players and ciliary cargoes may differ according to the cell-type, the experimental conditions used and the stage of the cilia cycle (formation, elongation, or maintenance). Therefore, expression analysis and interactome profiles of autophagy players and co-factors in the same experimental conditions, could help understanding why the same biological process acts differently, even in the same cell type, and could provide information about the context-dependent role of autophagy in the control of ciliogenesis.
The ciliary compartment may represent a specialized site for effective autophagy control in response to cilia-mediated extracellular stimuli (e.g., fluid flow, pressure, and nutritional needs). Thus, cilia-mediated autophagy could control intracellular metabolic responses and govern physiological adaptations of organs during differentiation and development. On the flip side of the same coin, the autophagic-lysosomal pathway may influence ciliogenesis through the selective degradation of cilia regulators. summarize proteins and molecules with a role both in autophagy and ciliogenesis.
Table 1. Proteins involved in autophagy and acting as positive modulators of ciliogenesis.
Table 2. Proteins involved in autophagy and acting as negative modulators of ciliogenesis.
Table 3. Chemicals modulating autophagy and acting on ciliogenesis.
Novel players in the cilia-autophagy game: lysosomes and mitochondria
Lysosome-cilia dual crosstalk
Besides autophagic proteins, lysosomal proteins are also involved in ciliogenesis, and conversely, ciliary proteins are involved in lysosomal functions. An example is the lysosomal v-ATPase accessory protein ATP6AP1 (ATPase H+ transporting accessory protein 1). Indeed, it has been shown that depletion of both maternal and zygotic atp6ap1b expression in zebrafish reduces the size of the Kupffer<apos;>s vesicle (the L-R organizer in fish), alters cilia length and disrupts the establishment of proper L-R asymmetry in early developmental stages [Citation90]. In addition, SNX10 (sorting nexin 10), a phosphoinositide-binding protein implicated in cargo sorting in the endosomal/lysosomal pathway, is required for the formation of primary cilia [Citation91]. Mechanistically, SNX10 interacts with the v-ATPase complex and targets it to the centrosome where both synergistically regulate the ciliary trafficking of RAB8A, a critical regulator of ciliary membrane extension [Citation91]. Finally, it has been reported that the lysosomal anion transporter SLC26A2 localizes to the cilia of human efferent duct cells where it may have a role in male fertility [Citation92].
Finetti et al. recently showed that IFT20 is required for the correct targeting of lysosomal hydrolases, by regulating mannose-6-phosphate trafficking. IFT20-deficient cells display an increased lysosomal size and impairment to CTSB (cathepsin B) activity, suggesting lysosomal dysfunction. Furthermore, the authors observed activation of TFEB (transcription factor EB), a master regulator of lysosome biogenesis, which likely compensates for lysosome dysfunction in IFT20-deficient cells. Importantly, the lysosome-related function of IFT20 is shared by non-ciliated and ciliated cells [Citation93].
Crosstalk between mitochondria and cilia
Mitochondria are involved in bioenergy production, biomolecule synthesis and cell death regulation. Controlled levels of reactive oxygen species (ROS) are constantly produced by mitochondria and act as important signaling molecules [Citation94]. Mitochondria continuously undergo balanced fission and fusion events, which reflect mitochondrial dynamics. These processes highly affect mitochondrial functions, as well as their morphology and are under tight control to maintain proper mitochondrial homeostasis. The main mitochondrial quality control processes include mitophagy (selective mitochondrial autophagy) and mitochondrial biogenesis [Citation95]. Thus, mitochondrial quality control plays a key role in the cellular stress responses, as well as during cancer progression and its associated cellular metabolic shift [Citation95].
Mitochondrial proteins have recently been found to also localize to the ciliary basal bodies and the transition zone, with either demonstrated or suggested roles in ciliogenesis. An example is provided by XPNPEP3/NPHPL1 and SLC41A1/NPHPL2 proteins, which localize to mitochondria and modulate cilia function, and when mutated, result in nephronophthisis [Citation96–98]. Another example of cilia-mitochondria potential interplay is given by the localization to centrosomes of the voltage-dependent anion channel family members VDAC1, VDAC2 and VDAC3, which are porins present in the mitochondrial outer membrane that share non-redundant functions in ciliogenesis. VDAC1 and VDAC3 are found on mother centrioles, while VDAC2 localizes to centriolar satellites. VDAC3 modulates centriole assembly by recruiting TTK/MPS1 (TTK protein kinase), an important regulator of centriole assembly, to centrosomes [Citation99]. The VDAC3-TTK module promotes ciliary disassembly during cell cycle entry and suppresses cilia assembly in proliferating cells [Citation100]. Moreover, VDAC1 negatively regulates ciliogenesis, since its downregulation leads to inappropriate ciliogenesis in cycling RPE cells, and its overexpression suppresses cilia formation [Citation100,Citation101]. Instead, VDAC2 appears to promote ciliogenesis [Citation100,Citation101]. Whether VDACs levels in centrosomes are controlled by mitochondria is an open question.
Recently, it was shown in cancer cells that hypoxia induces cleavage of VDAC1 to VDAV1-▲C, which reprogrammes cells by increasing glycolysis and mitochondrial respiration thus favoring cell survival [Citation102]. Intriguingly, VDAV1-▲C seems to lose its putative interaction site with tubulin and microtubules, which is thought to bring about repression of ciliogenesis, indicating a link between cancer-associated metabolic adaptation and cilia impairment [Citation102]. In summary, VDAC emerges as a potential player in the crosstalk between mitochondria and ciliogenesis, and eventually cell proliferation and survival. Nevertheless, more studies are needed to shed light on this crosstalk.
A connection between mitochondria and motile cilia has also been described. Indeed, Burkhalter et al. showed that mitochondria influence cilia-dependent processes such as cilia length and motility in L-R asymmetry establishment in zebrafish embryos. Along these lines, whole exome analysis of heterotaxic patients revealed an increased burden of rare damaging variants in mitochondria-associated genes [Citation103]. Another link between motile cilia and mitochondria came from work of Hofherr et al., who identified the Ca2+-regulated mitochondrial ATP-Mg/Pi SLC25A25 (solute carrier family 25 member 25), which acts downstream of the ciliary PKD2/TRPP2 (polycystin 2, transient receptor potential cation channel) as an essential component of cilia-dependent pathways controlling the determination of L-R asymmetry in zebrafish [Citation104]. This connection needs to be confirmed in other animal models but could provide a missing link for the molecular mechanisms behind the first asymmetric gene expression that occurs at the L-R organizer in zebrafish.
Crosstalk between mitochondria and the autophagy-cilia axis
Despite the scarcity of published data, an inter-dependency between ciliogenesis, mitochondrial function and autophagy seems to exist and be related with cellular stress responses. Bae and colleagues showed that mitochondrial stress induced by uncoupling agents (inhibitors of respiratory complexes) or depletion of the pro-fusion OPA1 gene, which induces fission, stimulates ciliary growth in neuronal SH-SY5Y cells, thus demonstrating a novel regulatory function for mitochondria dynamics in ciliogenesis [Citation105]. Moreover, these authors showed that mitochondrial stress-induced ciliogenesis is dependent on ROS generation and AMPK activity, triggering autophagy by inhibiting the MTOR pathway (). Likewise, blockade of mitochondria-mediated autophagy by silencing Autophagy-Related 5 (Atg5) expression, prevents mitochondrial stress-related cilia elongation [Citation105]. In the same study, it was shown that autophagy induced by mitochondrial stress increases the formation of primary cilia through enhancement of OFD1 degradation under nutrient-rich conditions () [Citation105]. In addition, treatment of mice with the mitochondrial toxin 1-methyl-4-phenylpyridinium (MPTP) elicits ciliary elongation and autophagy in dopaminergic neurons of the substantia nigra. Indeed, blockade of cilia formation in these neurons attenuates MPTP-induced autophagy and facilitates dopaminergic neuron loss and motor disability, suggesting a cytoprotective effect for the mitochondrial stress-induced ciliogenesis [Citation105]. Accordingly, a transcriptomic study revealed that whenever the mitochondrial biogenesis control gene TFAM (transcription factor A, mitochondrial) is depleted, there is an upregulation of cilia and CFAP65 (cilia and flagella associated protein 65)[Citation106], which may indicate once more that some sort of mitochondrial stress promotes ciliogenesis. In summary, mitochondrial stress appears to promote ciliogenesis, which in turn is a cytoprotective process.
Figure 4. Mitochondria-autophagy-cilia crosstalk models. (a) Mitochondrial stress, such as depletion of the pro-fusion OPA1 gene, and ROS generation stimulate autophagy by activating AMPK and inhibiting the MTOR pathway. Mitochondrial stress-induced autophagy acts as a cytoprotective process by promoting ciliogenesis through enhanced degradation of OFD1. (b) Shear stress induced by biological fluids stimulates AMPK-dependent selective degradation of lipid droplets by autophagy (lipophagy), contributing to the production of fatty acids that provide mitochondrial substrates to generate ATP and produce energy. Moreover, AMPK stimulates mitochondrial biogenesis, which contributes to ATP production. The concomitant mitochondrial biogenesis and lipophagy contribute to control ATP-dependent cellular mechanisms.
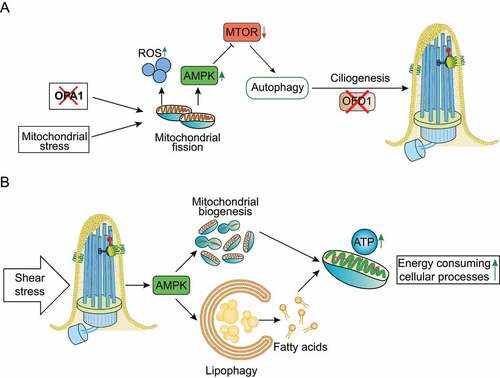
However, cilia-autophagy-mitochondria communication is not limited to mitochondrial stress/autophagy-mediated ciliogenesis. Miceli and colleagues recently showed that shear stress induced by biological fluids in KECs stimulates AMPK-mediated lipophagy (i.e., selective degradation of lipid droplets by autophagy), contributing to the production of fatty acids that provide mitochondrial substrates to generate ATP and produce energy (). Moreover, in KECs fluid flow stimulates AMPK-mediated mitochondrial biogenesis as assessed by the increased levels of mitochondrial proteins and DNA, and these flow-induced processes are dependent on primary cilia (). Finally, the concomitant cilia-dependent lipophagy and mitochondrial biogenesis contribute to ATP production, which is required to support energy-consuming cellular processes such as glucose reabsorption, gluconeogenesis and cytoskeletal remodeling in renal epithelial cells, demonstrating that the primary cilium, autophagy and mitochondrial metabolism are strictly interconnected () [Citation69,Citation70].
One can speculate that ciliogenesis acts as a cytoprotective process as it is stimulated by cellular stress. In fact, in response to a certain stress (below the threshold of causing cell death) cells stop proliferation (senescence), which may be associated with ciliogenesis. Likewise, ciliogenesis could also be involved in the activation of endogenous mechanisms of defense leading to tolerance or a preconditioning state. Nevertheless, whether ciliogenesis is a cytoprotective response against stress needs further investigation.
In glial cells primary cilia regulate the survival of astrocytes under unfavorable conditions such as starvation. In contrast, disruption of ciliogenesis associated with increased cell proliferation occurs in human astrocytoma/glioblastoma cells. Thus, one can speculate that ciliogenesis disruption might be associated with the reduction of mitochondrial metabolism and stimulation of autophagy (mitophagy), presenting increased levels of glycolysis, i.e., the Warburg effect.
Autophagy- and cilia-related genetic diseases
Recently, cilia function and autophagy have been recently functionally linked with a possible impacts on human health and disease [Citation38,Citation107]. At least some of the clinical manifestations observed in ciliopathies may be the result of perturbed autophagy and, conversely, cilia dysfunction could play a role in the increasing number of autophagy-related disorders.
Below, we present examples of pathways, genes and diseases associated with both cilia abnormalities and autophagic defects ().
Table 4. Ciliary- and autophagy-associated genes and human diseases.
Oral-facial-digital syndrome 1 (OFD type I)
OFD type I syndrome is characterized by renal, hepatic and pancreatic cysts, skeletal defects, central nervous system involvement (malformations and intellectual disabilities) and orofacial abnormalities [Citation26,Citation108]. As discussed above, the OFD1 protein was recently shown to act as a novel autophagy receptor controlling autophagosome biogenesis [Citation78]. In particular, it has been reported that dysregulated autophagy is associated with the pathogenesis of renal cystic disease in a mutant model of OFD type I syndrome in which autophagy modulation improves renal cystogenesis, suggesting that autophagy modulation might be a therapeutic target for renal cystic disease [Citation36,Citation78,Citation79,Citation109].
Autosomal dominant polycystic kidney disease (ADPKD)
PKD1 (polycystin 1, transient receptor potential channel interacting) and PKD2 localize to primary cilia in kidneys [Citation110,Citation111] where they are thought to form a complex that mediates mechanosensation through sensing cilia bending in response to fluid flow and triggering a calcium influx [Citation112]. Mutations in PKD1 or PKD2 are associated with ADPKD, characterized by the cardinal manifestations of renal cysts, liver cysts, and intracranial aneurysm. In 2006, it was shown for the first time that the MTOR pathway, the major suppressor of catabolic processes such as autophagy, is inappropriately activated in renal cysts in ADPKD patients and murine models [Citation113,Citation114]. Treatment with the MTOR inhibitor rapamycin was shown to reduce renal cystogenesis in murine models [Citation113,Citation114]. However, treatment with sirolimus, an MTOR inhibitor, cannot halt polycystic kidney growth in ADPKD patients [Citation115,Citation116].
Several studies firmly linked PC1 and PC2 with autophagy regulation [Citation67,Citation117–119]. For example, in kidney epithelial cells and in response to shear stress, autophagy is stimulated not only by the MTORC1-AMPK pathway, as mentioned above, but also by calcium influx dependent on PC2 [Citation67]. However, the role of PC2-dependent autophagy remains to be clarified since it is not involved in cell size regulation, as for MTORC1-AMPK dependent autophagy. Moreover, PC2 regulates autophagy independently of the location in the primary cilium (for a review see [Citation120]).
In 2011, altered autophagy was demonstrated in rat and murine models of PKD [Citation121]. Moreover, defective glucose metabolism in ADPKD has been linked with abnormal autophagy [Citation122] and increased autophagy has been shown to improve the renal cystic phenotype in pkd1 and pkd2 deficient zebrafish models [Citation123]. The hypothesis that suppression of autophagy may play a role in cyst formation and growth in ADPKD [Citation124] has been put forward. However, the experimental evidence to support the role of autophagy in PKD remains limited and controversial.
Focal malformations of cortical development (FMCDs)
Brain somatic mutations in MTOR gene are associated with FMCDs, which are major etiologies of pediatric intractable epilepsies exhibiting cortical dyslamination [Citation125]. Park et al. showed that neuronal ciliogenesis is defective in FMCDs due to loss of autophagy-mediated degradation of the centriolar satellite portion of the OFD1 protein [Citation126]. These findings were confirmed in murine models and in brain tissues from patients. Furthermore, the authors demonstrated that disruption of autophagy-mediated ciliogenesis leads to FMCD by abrogating Wnt signaling transduction [Citation126].
Tuberous sclerosis complex (TSC)
TSC is an autosomal dominant multisystemic disease caused by mutation in either the TSC1 [Citation127] or TSC2 [Citation128] tumor suppressor genes and characterized by a variety of symptoms such as nonmalignant brain or heart tumors, kidney and lung cysts, as well as neurocognitive defects (reviewed in [Citation129]). The TSC1 and TSC2 gene products, also known as hamartin and tuberin respectively, form a stable functional complex in vivo [Citation130] and impact MTOR mediated downstream signaling [Citation131,Citation132], including autophagy [Citation133,Citation134]. Indeed, it was shown in several studies that TSC1 and TSC2 regulate primary cilia length, structure and function [Citation135–138] (reviewed in [Citation139]). Additionally, a recent study demonstrated that brains of TSC patients and murine models have reduced ciliated neurons and that MTORC1 activity inversely correlates with ciliation in TSC1- and TSC2-deficient neurons, which is restored by rapamycin treatment. The authors also identified inhibitors of the heat shock protein that suppress MTORC1 activity and rescue ciliation, leading to the identification of the heat-shock machinery as a druggable signaling node to restore MTORC1 activity and cilia impairment due to loss of TSC1 and TSC2 [Citation140].
Joubert syndrome (JBTS)
Joubert syndrome is a ciliopathy characterized mainly by a distinctive cerebellar and brain stem malformation called the molar tooth sign, hypotonia, retinal involvement and developmental delay. INPP5E, which is one of the genes mutated in JBTS [Citation141] encodes an PtdIns(1,4,5)P3 5-phosphatase localized to primary cilia, where it controls the ciliary PIPs, which impacts on the proper trafficking of primary cilia-associated signaling proteins and HH transduction. As a consequence of INPP5E inactivation, cells display shorter cilia [Citation141] and suppression of the cilia-mediated HH signaling pathway [Citation142,Citation143]. Hasegawa et al. demonstrated that INPP5E also localizes to lysosomes and is required for autophagosome-lysosome fusion in a cilia-independent manner [Citation144]. Similarly, the same authors showed that patients with INPP5E mutations affecting the phosphatase activity of the enzyme are associated with impaired autophagy [Citation144]. Future studies should focus on determining whether autophagy defects underlie some of the clinical manifestations observed in JBTS.
Lowe oculocerebrorenal syndrome and dent-2 disease
OCRL/OCRL1 encodes a PtdIns(4,5)P2 5-phosphatase, which localizes to basal bodies and along axonemes, and plays a role in cilia biogenesis [Citation145]. OCRL is mutated in Lowe oculocerebrorenal syndrome and Dent-2 disease, two rare X-linked conditions [Citation146] in which patient fibroblasts display shorter cilia [Citation145,Citation147,Citation148]. De Leo and colleagues demonstrated that OCRL plays a direct functional role in autophagy, since it is recruited to lysosomes and is required for autophagosome-lysosome fusion [Citation149]. Moreover, loss of OCRL catalytic activity causes accumulation of autophagosomes and lysosomal dysfunction in cells isolated from Lowe syndrome patient cells [Citation149].
Birt-Hogg-Dubé (BHD) syndrome
FLCN (folliculin) gene mutations are associated with BHD syndrome, which predisposes patients to benign hair follicle tumors, pneumothorax, and renal cancer. FLCN encodes a GTPase-activating protein (GAP) localized on lysosomal membranes that plays a key role in the cellular response to amino acids through the MTORC1 signaling cascade [Citation150–154]. FLCN GAP activity is inhibited during starvation and activated in the presence of nutrients, stimulating MTORC1 [Citation155]. In addition, FLCN promotes autophagy following phosphorylation by ULK1 and physical interaction with GABARAP [Citation156]. In clear cell tumors from a BHD patient, loss of FLCN was shown to increase GABARAP and LC3 puncta, as well as SQSTM1/p62 levels, demonstrating that autophagy is impaired in BHD-associated renal tumors. Besides its role in nutrient sensing and autophagy, FLCN also localizes to primary cilia and its inactivation results in impaired ciliogenesis and dysregulation of cilia-dependent WNT and PCP signaling pathways [Citation30]. Recently, Zhong and colleagues showed that FLCN is part of a flow sensory mechanism that regulates MTORC1 through primary cilia [Citation157]. In response to flow stress, FLCN recruits STK11/LKB1 to primary cilia for the activation of AMPK localized to basal bodies, resulting in MTORC1 downregulation. In cells depleted for FLCN, STK11/LKB1 fails to accumulate in primary cilia and AMPK at the basal bodies remains inactive, thus nullifying the inhibitory effect of flow stress on MTORC1 activity [Citation157].
Other conditions associated with autophagic defects and possibly involving also cilia dysfunction include Huntington disease (HD) (reviewed in [Citation81,Citation158–160] and disorders associated with mutations in PTEN (phosphatase and tensin homolog) gene [Citation161–164]. However, for these diseases a clear causal relation with cilia dysfunction has not been clearly established.
Conclusions and future perspectives
Experimental evidence suggests that primary cilia represent a specialized site for effective autophagy control and that selective autophagic degradation removes positive and/or negative regulators of ciliogenesis, thus influencing cilia formation and elongation.
However, contradictory findings have been reported since autophagic degradation can exert an opposite role on cilia biology in a context-dependent manner. One critical point would be to establish standard culture conditions and methodologies to compare results from different laboratories worldwide. Moreover, the picture is getting more complex as new players have arisen, such as mitochondria and lysosomes, although our knowledge of them is still very limited.
The list of genes associated with ciliopathies or ciliary dysfunction and encoding proteins controlling autophagy keeps growing, suggesting far-reaching implications for cilium- and autophagy-related disorders. It is tempting to speculate that modulation of autophagy might represent a potential therapeutic opportunity to treat ciliopathies and, conversely, analysis of ciliogenesis might represent a good readout for therapeutic approaches involving modulators of autophagy in diseases not currently associated with primary cilia dysfunction [Citation38].
Disclosure statement
The authors declare that they have no competing interests.
Additional information
Funding
References
- Mizushima N. A brief history of autophagy from cell biology to physiology and disease. Nat Cell Biol. 2018;20(5):521–527.
- He LQ, Lu JH, Yue ZY. Autophagy in ageing and ageing-associated diseases. Acta Pharmacol Sin. 2013;34(5):605–611.
- Jiang P, Mizushima N. Autophagy and human diseases. Cell Res. 2014;24(1):69–79.
- Stamatakou E, Wróbel L, Hill SM, et al. Mendelian neurodegenerative disease genes involved in autophagy. Cell Discov. 2020;6(1):24. DOI:10.1038/s41421-020-0158-y.
- Klionsky DJ, Eskelinen E-L, Deretic V. Autophagosomes, phagosomes, autolysosomes, phagolysosomes, autophagolysosomes … wait, I’m confused. Autophagy. 2014;10(4):549–551.
- Duve de C. Lysosomes revisited. Eur J Biochem. 1983;137(3):391–397.
- Melia TJ, Lystad AH, Simonsen A. Autophagosome biogenesis: from membrane growth to closure. J Cell Biol. 2020;219(6):e202002085.
- Condon KJ, Sabatini DM. Nutrient regulation of mTORC1 at a glance. J Cell Sci. 2019;132(21):0–2.
- King KE, Losier TT, Russell RC. Regulation of autophagy enzymes by nutrient signaling. Trends Biochem Sci. 2021;46(8):687–700.
- Johansen T, Lamark T. Selective Autophagy: ATG8 Family Proteins, LIR Motifs and Cargo Receptors. J Mol Biol. 2020;432(1):80–103.
- Stolz A, Ernst A, Dikic I. Cargo recognition and trafficking in selective autophagy. Nat Cell Biol. 2014;16(6):495–501.
- Gubas A, Dikic I. A guide to the regulation of selective autophagy receptors. FEBS J. 2022;289(1):75–89.
- Satir P, Heuser T, Sale WS, et al. Basis for How Motile Cilia Beat. Bioscience. 2014;64(12):1073–1083.
- Woolley DM. Flagellar oscillation: a commentary on proposed mechanisms. Biol Rev Camb Philos Soc. 2010;85(3):453–470.
- Aprea I, Nöthe-Menchen T, Dougherty GW, et al. Motility of efferent duct cilia aids passage of sperm cells through the male reproductive system. Mol Hum Reprod. 2021;27(3):gaab009. DOI:10.1093/molehr/gaab009.
- Little RB, Norris DP. Right, left and cilia: how asymmetry is established. Semin Cell Dev Biol. 2021;110:11–18.
- Kobayashi T, Dynlacht BD. Regulating the transition from centriole to basal body. J Cell Biol. 2011;193(3):435–444.
- Conduit PT, Wainman A, Raff JW. Centrosome function and assembly in animal cells. Nat Rev Mol Cell Biol. 2015;16(10):611–624.
- Gonçalves J, Pelletier L. The Ciliary Transition Zone: finding the Pieces and Assembling the Gate. Mol Cells. 2017;40(4):243–253.
- Ishikawa H, Marshall WF. Intraflagellar Transport and Ciliary Dynamics. Cold Spring Harb Perspect Biol. 2017;9(3):a021998.
- Shakya S, Westlake CJ. Recent advances in understanding assembly of the primary cilium membrane. Fac Rev. 2021;10:10.
- Wang L, Dynlacht BD. The regulation of cilium assembly and disassembly in development and disease. Development. 2018;145(18):18.
- Reiter JF, Leroux MR. Genes and molecular pathways underpinning ciliopathies. Nat Rev Mol Cell Biol. 2017;18(9):533–547.
- Bergmann C, Guay-Woodford LM, Harris PC, et al. Polycystic kidney disease. Nat Rev Dis Prim. 2018;4(1):50. DOI:10.1038/s41572-018-0047-y.
- Franco B, Thauvin-Robinet C. Update on oral-facial-digital syndromes (OFDS). Cilia. 2016;5(1):12.
- Morleo M, Franco B, Type OFD. I syndrome: lessons learned from a rare ciliopathy. Biochem Soc Trans. 2020;48(5):1929–1939.
- Pezzella N, Bove G, Tammaro R, et al. OFD1: one gene, several disorders. Am J Med Genet C Semin Med Genet. 2022; Online ahead of print. DOI:10.1002/ajmg.c.31962.
- Parisi MA. The molecular genetics of Joubert syndrome and related ciliopathies: the challenges of genetic and phenotypic heterogeneity. Trans Sci Rare Dis. 2019;4(1–2):25–49.
- Barker AR, Thomas R, Dawe HR. Meckel-Gruber syndrome and the role of primary cilia in kidney, skeleton, and central nervous system development. Organogenesis. 2014;10(1):96–107.
- Luijten MN, Basten SG, Claessens T, et al. Birt-Hogg-Dube syndrome is a novel ciliopathy. Hum Mol Genet. 2013;22(21):4383–4397. DOI:10.1016/j.cell.2019.10.036.
- Braun DA, Hildebrandt F. Hildebrandt F. Ciliopathies. Cold Spring Harb Perspect Biol. 2017;9(3):a028191.
- Cao M, Zhong Q. Cilia in autophagy and cancer. Cilia. 2015;5(1):4.
- Yun C, Lee S. The Roles of Autophagy in Cancer. Int J Mol Sci. 2018;19(11):3466.
- Wang B, Liang Z, Liu P. Functional aspects of primary cilium in signaling, assembly and microenvironment in cancer. J Cell Physiol. 2021;236(5):3207–3219.
- Wallmeier J, Nielsen KG, Kuehni CE, et al. Motile ciliopathies. Nat Rev Dis Prim. 2020;6(1):77. DOI:10.1038/s41572-020-0209-6.
- Franco B, Morleo M. The role of OFD1 in selective autophagy. Mol Cell Oncol. 2021;8(3):1903291.
- Pampliega O, Cuervo AM. Autophagy and primary cilia: dual interplay. Curr Opin Cell Biol. 2016;39:1–7.
- Morleo F. The Autophagy-Cilia Axis: an Intricate Relationship. Cells. 2019;8(8):905.
- Yamamoto Y, Chino H, Tsukamoto S, et al. NEK9 regulates primary cilia formation by acting as a selective autophagy adaptor for MYH9/myosin IIA. Nat Commun. 2021;12(1):3292. DOI:10.1038/s41467-021-23599-7.
- Tang Z, Lin MG, Stowe TR, et al. Autophagy promotes primary ciliogenesis by removing OFD1 from centriolar satellites. Nature. 2013;502(7470):254–257. DOI:10.1038/nature12606.
- Singla V, Romaguera-Ros M, Garcia-Verdugo JM, et al. Ofd1, a human disease gene, regulates the length and distal structure of centrioles. Dev Cell. 2010;18(3):410–424. DOI:10.1016/j.devcel.2009.12.022.
- Ferrante MI, Zullo A, Barra A, et al. Oral-facial-digital type I protein is required for primary cilia formation and left-right axis specification. Nat Genet. 2006;38(1):112–117. DOI:10.1038/ng1684.
- Pampliega O, Orhon I, Patel B, et al. Functional interaction between autophagy and ciliogenesis. Nature. 2013;502(7470):194–200. DOI:10.1038/nature12639.
- Boukhalfa A, Miceli C, Ávalos Y, et al. Interplay between primary cilia, ubiquitin-proteasome system and autophagy. Biochimie. 2019;166:286–292.
- Hubbert C, Guardiola A, Shao R, et al. HDAC6 is a microtubule-associated deacetylase. Nature. 2002;417(6887):455–458. DOI:10.1038/417455a.
- Lee J-Y, Koga H, Kawaguchi Y, et al. HDAC6 controls autophagosome maturation essential for ubiquitin-selective quality-control autophagy. EMBO J. 2010;29(5):969–980. DOI:10.1038/emboj.2009.405.
- Lam HC, Cloonan SM, Bhashyam AR, et al. Histone deacetylase 6-mediated selective autophagy regulates COPD-associated cilia dysfunction. J Clin Invest. 2013;123(12):5212–5230. DOI:10.1172/JCI69636.
- Spektor A, Tsang WY, Khoo D, et al. Cep97 and CP110 Suppress a Cilia Assembly Program. Cell. 2007;130(4):678–690. DOI:10.1016/j.cell.2007.06.027.
- Bettencourt-Dias M, Carvalho-Santos Z. Double life of centrioles: CP110 in the spotlight. Trends Cell Biol. 2008;18(1):8–11.
- Liu M, Zhang W, Li M, et al. NudCL2 is an autophagy receptor that mediates selective autophagic degradation of CP110 at mother centrioles to promote ciliogenesis. Cell Res. 2021;31(11):1199–1211. DOI:10.1038/s41422-021-00560-3.
- Arora K, Lund JR, Naren NA, et al. AC6 regulates the microtubule-depolymerizing kinesin KIF19A to control ciliary length in mammals. J Biol Chem. 2020;295(42):14250–14259. DOI:10.1074/jbc.RA120.013703.
- Leopold PL, O’Mahony MJ, Lian XJ, et al. Smoking is associated with shortened airway cilia. PLoS One. 2009;4(12):e8157. DOI:10.1371/journal.pone.0008157.
- Hessel J, Heldrich J, Fuller J, et al. Intraflagellar transport gene expression associated with short cilia in smoking and COPD. PLoS One. 2014;9(1):e85453. DOI:10.1371/journal.pone.0085453.
- Bottier M, Thomas KA, Dutcher SK, et al. How Does Cilium Length Affect Beating? Biophys J. 2019;116(7):1292–1304. DOI:10.1016/j.bpj.2019.02.012.
- Pintado P, Sampaio P, Tavares B, et al. Dynamics of cilia length in left–right development. R Soc Open Sci. 2017;4(3):161102. DOI:10.1098/rsos.161102.
- Niwa S, Nakajima K, Miki H, et al. KIF19A Is a Microtubule-Depolymerizing Kinesin for Ciliary Length Control. Dev Cell. 2012;23(6):605–611. DOI:10.1016/j.devcel.2012.10.016.
- Moore BS, Stepanchick AN, Tewson PH, et al. Cilia have high cAMP levels that are inhibited by Sonic Hedgehog-regulated calcium dynamics. Proc Natl Acad Sci. 2016;113(46):549–551. DOI:10.1073/pnas.1602393113.
- Besschetnova TY, Kolpakova-Hart E, Guan Y, et al. Identification of Signaling Pathways Regulating Primary Cilium Length and Flow-Mediated Adaptation. Curr Biol. 2010;20(2):182–187. DOI:10.1016/j.cub.2009.11.072.
- Flaherty J, Feng Z, Peng Z, et al. Primary cilia have a length-dependent persistence length. Biomech Model Mechanobiol. 2020;19(2):445–460. DOI:10.1007/s10237-019-01220-7.
- Volinia S, Dhand R, Vanhaesebroeck B, et al. A human phosphatidylinositol 3-kinase complex related to the yeast Vps34p-Vps15p protein sorting system. EMBO J. 1995;14(14):3339–3348. DOI:10.1002/j.1460-2075.1995.tb07340.x.
- Lindmo K, Brech A, Finley KD, et al. The PI 3-kinase regulator Vps15 is required for autophagic clearance of protein aggregates. Autophagy. 2008;4(4):500–506. DOI:10.4161/auto.5829.
- Stoetzel C, Bär S, Craene De J-O, et al. A mutation in VPS15 (PIK3R4) causes a ciliopathy and affects IFT20 release from the cis-Golgi. Nat Commun. 2016;7(1):13586. DOI:10.1038/ncomms13586.
- Finetti F, Paccani SR, Rosenbaum J, et al. Intraflagellar transport: a new player at the immune synapse. Trends Immunol. 2011;32(4):139–145. DOI:10.1016/j.it.2011.02.001.
- Boukhalfa A, Roccio F, Dupont N, et al. The autophagy protein ATG16L1 cooperates with IFT20 and INPP5E to regulate the turnover of phosphoinositides at the primary cilium. Cell Rep. 2021;35(4):109045. DOI:10.1016/j.celrep.2021.109045.
- Boukhalfa A, Roccio F, Dupont N, et al. When the autophagy protein ATG16L1 met the ciliary protein IFT20. Autophagy. 2021;17(7):1791–1793. DOI:10.1080/15548627.2021.1935004.
- Zemirli N, Boukhalfa A, Dupont N, et al. The primary cilium protein folliculin is part of the autophagy signaling pathway to regulate epithelial cell size in response to fluid flow. Cell Stress. 2019;3(3):100–109. DOI:10.15698/cst2019.03.180.
- Orhon I, Dupont N, Zaidan M, et al. Primary-cilium-dependent autophagy controls epithelial cell volume in response to fluid flow. Nat Cell Biol. 2016;18(6):657–667. DOI:10.1038/ncb3360.
- Xiang W, Jiang T, Hao X, et al. Primary cilia and autophagy interaction is involved in mechanical stress mediated cartilage development via ERK/mTOR axis. Life Sci. 2019;218:308–313.
- Miceli C, Roccio F, Penalva-Mousset L, et al. The primary cilium and lipophagy translate mechanical forces to direct metabolic adaptation of kidney epithelial cells. Nat Cell Biol. 2020;22(9):1091–1102. DOI:10.1038/s41556-020-0566-0.
- Miceli C, Roccio F, Penalva-Mousset L, et al. Fluid flow-induced shear stress controls the metabolism of proximal tubule kidney epithelial cells through primary cilium-dependent lipophagy and mitochondria biogenesis. Autophagy. 2020;16(12):2287–2288. DOI:10.1080/15548627.2020.1823125.
- Jang J, Wang Y, Lalli MA, et al. Primary cilium-autophagy-Nrf2 (PAN) axis activation commits human embryonic stem cells to a neuroectoderm fate. Cell. 2016;165(2):410–420. DOI:10.1016/j.cell.2016.02.014.
- Boehlke C, Kotsis F, Patel V, et al. Primary cilia regulate mTORC1 activity and cell size through Lkb1. Nat Cell Biol. 2010;12(11):1115–1122. DOI:10.1038/ncb2117.
- Boukhalfa A, Nascimbeni AC, Ramel D, et al. PI3KC2α-dependent and VPS34-independent generation of PI3P controls primary cilium-mediated autophagy in response to shear stress. Nat Commun. 2020;11(1):294. DOI:10.1038/s41467-019-14086-1.
- Franco I, Gulluni F, Campa CC, et al. PI3K class II α controls spatially restricted endosomal PtdIns3P and Rab11 activation to promote primary cilium function. Dev Cell. 2014;28(6):647–658. DOI:10.1016/j.devcel.2014.01.022.
- WuDunn D. Mechanobiology of trabecular meshwork cells. Exp Eye Res. 2009;88(4):718–723.
- Shim MS, Nettesheim A, Hirt J, et al. The autophagic protein LC3 translocates to the nucleus and localizes in the nucleolus associated to NUFIP1 in response to cyclic mechanical stress. Autophagy. 2020;16(7):1248–1261. DOI:10.1080/15548627.2019.1662584.
- Hirt J, Liton PB. Autophagy and mechanotransduction in outflow pathway cells. Exp Eye Res. 2017;158:146–153.
- Morleo M, Brillante S, Formisano U, et al. Regulation of autophagosome biogenesis by OFD1mediated selective autophagy. EMBO J. 2021;40(4):4. DOI:10.15252/embj.2020105120.
- Morleo M, Franco B. The OFD1 protein is a novel player in selective autophagy: another tile to the cilia/autophagy puzzle. Cell Stress. 2021;5(3):33–36.
- Hoang-Minh LB, Deleyrolle LP, Nakamura NS, et al. PCM1 depletion inhibits glioblastoma cell ciliogenesis and increases cell death and sensitivity to temozolomide. Transl Oncol. 2016;9(5):392–402. DOI:10.1016/j.tranon.2016.08.006.
- Keryer G, Pineda JR, Liot G, et al. Ciliogenesis is regulated by a huntingtin-HAP1-PCM1 pathway and is altered in Huntington disease. J Clin Invest. 2011;121(11):4372–4382. DOI:10.1172/JCI57552.
- Joachim J, Razi M, Judith D, et al. Centriolar satellites control GABARAP ubiquitination and GABARAP-mediated autophagy. Curr Biol. 2017;27(14):2123–2136 e7. DOI:10.1016/j.cub.2017.06.021.
- Finetti F, Cassioli C, Cianfanelli V, et al. The intraflagellar transport protein IFT20 recruits ATG16L1 to early endosomes to promote autophagosome formation in T cells. Front Cell Dev Biol. 2021;9 :634003.
- Liu ZQ, Lee JN, Son M, et al. Ciliogenesis is reciprocally regulated by PPARA and NR1H4/FXR through controlling autophagy in vitro and in vivo. Autophagy. 2018;14(6):1011–1027. DOI:10.1080/15548627.2018.1448326.
- Kim ES, Shin JH, Park SJ, et al. Inhibition of autophagy suppresses sertraline-mediated primary ciliogenesis in retinal pigment epithelium cells. PLoS One. 2015;10(2):e0118190. DOI:10.1371/journal.pone.0118190.
- Bao Z, Huang W. Thioridazine promotes primary ciliogenesis in lung cancer cells through enhancing cell autophagy. Int J Clin Exp Med. 2017;10(9):13960–13969.
- Struchtrup A, Wiegering A, Stork B, et al. The ciliary protein RPGRIP1L governs autophagy independently of its proteasome-regulating function at the ciliary base in mouse embryonic fibroblasts. Autophagy. 2018;14(4):567–583. DOI:10.1080/15548627.2018.1429874.
- Wang S, Livingston MJ, Su Y, et al. Reciprocal regulation of cilia and autophagy via the MTOR and proteasome pathways. Autophagy. 2015;11(4):607–616. DOI:10.1080/15548627.2015.1023983.
- Iaconis D, Crina C, Brillante S, et al. The HOPS complex subunit VPS39 controls ciliogenesis through autophagy. Hum Mol Genet. 2020;29(6):1018–1029. DOI:10.1093/hmg/ddaa029.
- Gokey JJ, Dasgupta A, Amack JD. The V-ATPase accessory protein Atp6ap1b mediates dorsal forerunner cell proliferation and left–right asymmetry in zebrafish. Dev Biol. 2015;407(1):115–130.
- Chen Y, Wu B, Xu L, et al. A SNX10/V-ATPase pathway regulates ciliogenesis in vitro and in vivo. Cell Res. 2012;22(2):333–345. DOI:10.1038/cr.2011.134.
- Kujala M, Hihnala S, Tienari J, et al. Expression of ion transport-associated proteins in human efferent and epididymal ducts. Reproduction. 2007;133(4):775–784. DOI:10.1530/rep.1.00964.
- Finetti F, Cassioli C, Cianfanelli V, et al. The intraflagellar transport protein IFT20 controls lysosome biogenesis by regulating the post-Golgi transport of acid hydrolases. Cell Death Differ. 2020;27(1):310–328. DOI:10.1038/s41418-019-0357-y.
- Dan Dunn J, Alvarez LAJ, Zhang X, et al. Reactive oxygen species and mitochondria: a nexus of cellular homeostasis. Redox Biol. 2015;6:472–485.
- Pickles S, Vigié P, Youle RJ. Mitophagy and quality control mechanisms in mitochondrial maintenance. Curr Biol. 2018;28(4):R170–85.
- Sang L, Miller JJ, Corbit KC, et al. Mapping the NPHP-JBTS-MKS protein network reveals ciliopathy disease genes and pathways. Cell. 2011;145(4):513–528. DOI:10.1016/j.cell.2011.04.019.
- Garcia-Gonzalo FR, Corbit KC, Sirerol-Piquer MS, et al. A transition zone complex regulates mammalian ciliogenesis and ciliary membrane composition. Nat Genet. 2011;43(8):776–784. DOI:10.1038/ng.891.
- Williams CL, Li C, Kida K, et al. MKS and NPHP modules cooperate to establish basal body/transition zone membrane associations and ciliary gate function during ciliogenesis. J Cell Biol. 2011;192(6):1023–1041. DOI:10.1083/jcb.201012116.
- Pike AN, Fisk HA. Centriole assembly and the role of Mps1: defensible or dispensable? Cell Div. 2011;6(1):9.
- Majumder S, Fisk HA. VDAC3 and Mps1 negatively regulate ciliogenesis. Cell Cycle. 2013;12(5):849–858.
- Majumder S, Cash A, Fisk H. Non-overlapping distributions and functions of the VDAC family in ciliogenesis. Cells. 2015;4(3):331–353.
- Meyenberg Cunha-de Padua M, Fabbri L, Dufies M, et al. Evidences of a direct relationship between cellular fuel supply and ciliogenesis regulated by hypoxic VDAC1-ΔC. Cancers (Basel). 2020;12(11):3484. DOI:10.3390/cancers12113484.
- Burkhalter MD, Sridhar A, Sampaio P, et al. Imbalanced mitochondrial function provokes heterotaxy via aberrant ciliogenesis. J Clin Invest. 2019;129(7):2841–2855. DOI:10.1172/JCI98890.
- Hofherr A, Seger C, Fitzpatrick F, et al. The mitochondrial transporter SLC25A25 links ciliary TRPP2 signaling and cellular metabolism. PLOS Biol. 2018;16(8):e2005651. DOI:10.1371/journal.pbio.2005651.
- Bae J-E, Kang GM, Min SH, et al. Primary cilia mediate mitochondrial stress responses to promote dopamine neuron survival in a Parkinson’s disease model. Cell Death Dis. 2019;10(12):952. DOI:10.1038/s41419-019-2184-y.
- Lee WR, Na H, Lee SW, et al. Transcriptomic analysis of mitochondrial TFAM depletion changing cell morphology and proliferation. Sci Rep. 2017;7(1):17841. DOI:10.1038/s41598-017-18064-9.
- Orhon I, Dupont N, Pampliega O, et al. Autophagy and regulation of cilia function and assembly. Cell Death Differ. 2015;22(3):389–397. DOI:10.1038/cdd.2014.171.
- Macca M, Franco B. The molecular basis of oral-facial-digital syndrome, type 1. Am J Med Genet C Semin Med Genet. 2009;151(4):318–325.
- Zullo A, Iaconis D, Barra A, et al. Kidney-specific inactivation of Ofd1 leads to renal cystic disease associated with upregulation of the mTOR pathway. Hum Mol Genet. 2010;19(14):2792–2803. DOI:10.1093/hmg/ddq180.
- Pazour GJ, San Agustin JT, Follit JA, et al. Polycystin-2 localizes to kidney cilia and the ciliary level is elevated in orpk mice with polycystic kidney disease. Curr Biol. 2002;12(11):R378–80. DOI:10.1016/S0960-9822(02)00877-1.
- Yoder BK, Hou X, Guay-Woodford LM. The polycystic kidney disease proteins, polycystin-1, polycystin-2, polaris, and cystin, are co-localized in renal cilia. J Am Soc Nephrol. 2002;13(10):2508–2516.
- Nauli SM, Alenghat FJ, Luo Y, et al. Polycystins 1 and 2 mediate mechanosensation in the primary cilium of kidney cells. Nat Genet. 2003;33(2):129–137. DOI:10.1038/ng1076.
- Shillingford JM, Murcia NS, Larson CH, et al. The mTOR pathway is regulated by polycystin-1, and its inhibition reverses renal cystogenesis in polycystic kidney disease. Proc Natl Acad Sci U S A. 2006;103(14):5466–5471. DOI:10.1073/pnas.0509694103.
- Boletta A. Emerging evidence of a link between the polycystins and the mTOR pathways. Pathogenetics. 2009;2(1):6.
- Serra AL, Poster D, Kistler AD, et al. Sirolimus and kidney growth in autosomal dominant polycystic kidney disease. N Engl J Med. 2010;363(9):820–829. DOI:10.1056/NEJMoa0907419.
- Kim HJ, Edelstein CL. Mammalian target of rapamycin inhibition in polycystic kidney disease: from bench to bedside. Kidney Res Clin Pr. 2012;31(3):132–138.
- Cebotaru V, Cebotaru L, Kim H, et al. Polycystin-1 negatively regulates Polycystin-2 expression via the aggresome/autophagosome pathway. J Biol Chem. 2014;289(10):6404–6414. DOI:10.1074/jbc.M113.501205.
- Peña-Oyarzun D, Rodriguez-Peña M, Burgos-Bravo F, et al. PKD2/polycystin-2 induces autophagy by forming a complex with BECN1. Autophagy. 2021;17(7):1714–1728. DOI:10.1080/15548627.2020.1782035.
- Peintner L, Venkatraman A, Waeldin A, et al. Loss of PKD1/polycystin-1 impairs lysosomal activity in a CAPN (calpain)-dependent manner. Autophagy. 2021;17(9):2384–2400. DOI:10.1080/15548627.2020.1826716.
- Peña-Oyarzun D, Batista-Gonzalez A, Kretschmar C, et al. New emerging roles of Polycystin-2 in the regulation of autophagy. Int Rev Cell Mol Biol. 2020;354:165–186.
- Belibi F, Zafar I, Ravichandran K, et al. Hypoxia-inducible factor-1alpha (HIF-1alpha) and autophagy in polycystic kidney disease (PKD). Am J Physiol Ren Physiol. 2011;300(5):F1235–43. DOI:10.1152/ajprenal.00348.2010.
- Rowe I, Chiaravalli M, Mannella V, et al. Defective glucose metabolism in polycystic kidney disease identifies a new therapeutic strategy. Nat Med. 2013;19(4):488–493. DOI:10.1038/nm.3092.
- Zhu P, Sieben CJ, Xu X, et al. Autophagy activators suppress cystogenesis in an autosomal dominant polycystic kidney disease model. Hum Mol Genet. 2017;26(1):158–172. DOI:10.1093/hmg/ddw376.
- Ravichandran K, Edelstein CL. Polycystic kidney disease: a case of suppressed autophagy? Semin Nephrol. 2014;34(1):27–33.
- Lim JS, Kim WI, Kang H-C-C, et al. Brain somatic mutations in MTOR cause focal cortical dysplasia type II leading to intractable epilepsy. Nat Med. 2015;21(4):395–400. DOI:10.1038/nm.3824.
- Park SM, Lim JS, Ramakrishina S, et al. Brain somatic mutations in MTOR disrupt neuronal ciliogenesis, leading to focal cortical dyslamination. Neuron. 2018;99(1):83–97 e7. DOI:10.1016/j.neuron.2018.05.039.
- Slegtenhorst van M, Hoogt de R, Hermans C, et al. Identification of the tuberous sclerosis gene TSC1 on chromosome 9q34. Science. 1997;277(5327):805–808. DOI:10.1126/science.277.5327.805.
- The European Chromosome 16 Tuberous Sclerosis Consortium. Identification and characterization of the tuberous sclerosis gene on chromosome 16. Cell. 1993;75(7):1305–1315. DOI:10.1016/0092-8674(93)90618-Z.
- Henske EP, Jóźwiak S, Kingswood JC, et al. Tuberous sclerosis complex. Nat Rev Dis Prim. 2016;2(1):16035. DOI:10.1038/nrdp.2016.35.
- Slegtenhorst van M. Interaction between hamartin and tuberin, the TSC1 and TSC2 gene products. Hum Mol Genet. 1998;7(6):1053–1057.
- Tee AR, Fingar DC, Manning BD, et al. Tuberous sclerosis complex-1 and −2 gene products function together to inhibit mammalian target of rapamycin (mTOR)-mediated downstream signaling. Proc Natl Acad Sci. 2002;99(21):13571–13576. DOI:10.1073/pnas.202476899.
- Inoki K, Li Y, Zhu T, et al. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat Cell Biol. 2002;4(9):648–657. DOI:10.1038/ncb839.
- Zhang J, Kim J, Alexander A, et al. A tuberous sclerosis complex signalling node at the peroxisome regulates mTORC1 and autophagy in response to ROS. Nat Cell Biol. 2013;15(10):1186–1196. DOI:10.1038/ncb2822.
- Papadakis M, Hadley G, Xilouri M, et al. Tsc1 (hamartin) confers neuroprotection against ischemia by inducing autophagy. Nat Med. 2013;19(3):351–357. DOI:10.1038/nm.3097.
- Hartman TR, Liu D, Zilfou JT, et al. The tuberous sclerosis proteins regulate formation of the primary cilium via a rapamycin-insensitive and polycystin 1-independent pathway. Hum Mol Genet. 2009;18(1):151–163. DOI:10.1093/hmg/ddn325.
- DiBella LM, Park A, Sun Z. Zebrafish Tsc1reveals functional interactions between the cilium and the TOR pathway. Hum Mol Genet. 2009;18(4):595–606.
- Rosengren T, Larsen LJ, Pedersen LB, et al. TSC1 and TSC2 regulate cilia length and canonical Hedgehog signaling via different mechanisms. Cell Mol Life Sci. 2018;75(14):2663–2680. DOI:10.1007/s00018-018-2761-8.
- Astrinidis A, Senapedis W, Henske EP. Hamartin, the tuberous sclerosis complex 1 gene product, interacts with polo-like kinase 1 in a phosphorylation-dependent manner. Hum Mol Genet. 2006;15(2):287–297.
- Mallela K, Kumar A. Role of TSC1 in physiology and diseases. Mol Cell Biochem. 2021;476(6):2269–2282.
- Di NA, Lenoël I, Winden KD, et al. Phenotypic screen with TSC-deficient neurons reveals heat-shock machinery as a druggable pathway for mTORC1 and reduced cilia. Cell Rep. 2020;31(12):107780. DOI:10.1016/j.celrep.2020.107780.
- Bielas SL, Silhavy JL, Brancati F, et al. Mutations in INPP5E, encoding inositol polyphosphate-5-phosphatase E, link phosphatidyl inositol signaling to the ciliopathies. Nat Genet. 2009;41(9):1032–1036. DOI:10.1038/ng.423.
- Chavez M, Ena S, Sande Van J, et al. Modulation of ciliary phosphoinositide content regulates trafficking and sonic hedgehog signaling output. Dev Cell. 2015;34(3):338–350. DOI:10.1016/j.devcel.2015.06.016.
- Garcia-Gonzalo FR, Phua SC, Roberson EC, et al. Phosphoinositides regulate ciliary protein trafficking to modulate hedgehog signaling. Dev Cell. 2015;34(4):400–409. DOI:10.1016/j.devcel.2015.08.001.
- Hasegawa J, Iwamoto R, Otomo T, et al. Autophagosome-lysosome fusion in neurons requires INPP5E, a protein associated with joubert syndrome. EMBO J. 2016;35(17):1853–1867. DOI:10.15252/embj.201593148.
- Luo N, West CC, Murga-Zamalloa CA, et al. OCRL localizes to the primary cilium: a new role for cilia in Lowe syndrome. Hum Mol Genet. 2012;21(15):3333–3344. DOI:10.1093/hmg/dds163.
- Conduit SE, Dyson JM, Mitchell CA. Inositol polyphosphate 5-phosphatases; new players in the regulation of cilia and ciliopathies. FEBS Lett. 2012;586(18):2846–2857.
- Coon BG, Hernandez V, Madhivanan K, et al. The Lowe syndrome protein OCRL1 is involved in primary cilia assembly. Hum Mol Genet. 2012;21(8):1835–1847. DOI:10.1093/hmg/ddr615.
- Montjean R, Aoidi R, Desbois P, et al. OCRL-mutated fibroblasts from patients with dent-2 disease exhibit INPP5B-independent phenotypic variability relatively to Lowe syndrome cells. Hum Mol Genet. 2015;24(4):994–1006. DOI:10.1093/hmg/ddu514.
- Leo De MG, Staiano L, Vicinanza M, et al. Autophagosome-lysosome fusion triggers a lysosomal response mediated by TLR9 and controlled by OCRL. Nat Cell Biol. 2016;18(8):839–850. DOI:10.1038/ncb3386.
- Baba M, Hong S-B, Sharma N, et al. Folliculin encoded by the BHD gene interacts with a binding protein, FNIP1, and AMPK, and is involved in AMPK and mTOR signaling. Proc Natl Acad Sci. 2006;103(42):15552–15557. DOI:10.1073/pnas.0603781103.
- Takagi Y, Kobayashi T, Shiono M, et al. Interaction of folliculin (birt-hogg-dubé gene product) with a novel Fnip1-like (FnipL/Fnip2) protein. Oncogene. 2008;27(40):5339–5347. DOI:10.1038/onc.2008.261.
- Petit CS, Roczniak-Ferguson A, Ferguson SM. Recruitment of folliculin to lysosomes supports the amino acid–dependent activation of rag GTPases. J Cell Biol. 2013;202(7):1107–1122.
- Tsun ZY, Bar-Peled L, Chantranupong L, et al. The folliculin tumor suppressor is a GAP for the RagC/D GTPases that signal amino acid levels to mTORC1. Mol Cell. 2013;52(4):495–505. DOI:10.1016/j.molcel.2013.09.016.
- Shen K, Rogala KB, Chou H-T, et al. Cryo-EM structure of the human FLCN-FNIP2-rag-ragulator complex. Cell. 2019;179(6):1319–1329.e8.
- Lawrence RE, Fromm SA, Fu Y, et al. Structural mechanism of a rag GTPase activation checkpoint by the lysosomal folliculin complex. Science. 2019;366(6468):971–977. DOI:10.1126/science.aax0364.
- Dunlop EA, Seifan S, Claessens T, et al. FLCN, a novel autophagy component, interacts with GABARAP and is regulated by ULK1 phosphorylation. Autophagy. 2014;10(10):1749–1760. DOI:10.4161/auto.29640.
- Zhong M, Zhao X, Li J, et al. Tumor suppressor folliculin regulates mTORC1 through primary cilia. J Biol Chem. 2016;291(22):11689–11697. DOI:10.1074/jbc.M116.719997.
- Barron JC, Hurley EP, Parsons MP. Huntingtin and the synapse. Front Cell Neurosci. 2021;15:689332.
- Rui YN, Xu Z, Patel B, et al. Huntingtin functions as a scaffold for selective macroautophagy. Nat Cell Biol. 2015;17(3):262–275. DOI:10.1038/ncb3101.
- Kaliszewski M, Knott AB, Bossy-Wetzel E. Primary cilia and autophagic dysfunction in Huntington’s disease. Cell Death Differ. 2015;22(9):9–24.
- Yehia L, Keel E, Eng C. The clinical spectrum of PTEN mutations. Annu Rev Med. 2020;71(1):103–116.
- Arico S, Petiot A, Bauvy C, et al. The tumor suppressor PTEN positively regulates macroautophagy by inhibiting the phosphatidylinositol 3-kinase/protein kinase B pathway. J Biol Chem. 2001;276(38):805–808. DOI:10.1074/jbc.C100319200.
- Niu Y, Sun W, Lu -J-J, et al. PTEN activation by DNA damage induces protective autophagy in response to cucurbitacin B in hepatocellular carcinoma cells. Oxid Med Cell Longev. 2016;2016:1–15.
- Shnitsar I, Bashkurov M, Masson GR, et al. PTEN regulates cilia through dishevelled. Nat Commun. 2015;6(1):8388.
- Peixoto E, Jin S, Thelen K, et al. HDAC6-dependent ciliophagy is involved in ciliary loss and cholangiocarcinoma growth in human cells and murine models. Am J Physiol Liver Physiol. 2020;318(6):G1022–33.
- Xu Q, Liu W, Liu X, et al. Type I collagen promotes primary cilia growth through down-regulating HDAC6-mediated autophagy in confluent mouse embryo fibroblast 3T3-L1 cells. J Biosci Bioeng. 2018;125(1):8–14. DOI:10.1016/j.jbiosc.2017.07.012.
- Lee J, Yi S, Kang YE, et al. Defective ciliogenesis in thyroid hürthle cell tumors is associated with increased autophagy. Oncotarget. 2016;7(48):79117–79130. DOI:10.18632/oncotarget.12997.
- Coene KLM, Roepman R, Doherty D, et al. OFD1 is mutated in X-linked joubert syndrome and interacts with LCA5-encoded lebercilin. Am J Hum Genet. 2009;85(4):465–481. DOI:10.1016/j.ajhg.2009.09.002.
- Conciliis de L, Marchitiello A, Wapenaar MC, et al. Characterization of Cxorf5 (71-7A), a novel human cDNA mapping to Xp22 and encoding a protein containing coiled-coila-helical domains. Genomics. 1998;51(2):243–250. DOI:10.1006/geno.1998.5348.
- Ferrante MI, Feather SA, Bulfone A, et al. Identification of the gene for oral-facial-digital type I syndrome. Am J Hum Genet. 2001;68(3):569–576. DOI:10.1086/318802.
- Bukowy-Bieryllo Z, Rabiasz A, Dabrowski M, et al. Truncating mutations in exons 20 and 21 of OFD1 can cause primary ciliary dyskinesia without associated syndromic symptoms. J Med Genet. 2019;56(11):769–777. DOI:10.1136/jmedgenet-2018-105918.
- Hannah WB, DeBrosse S, Kinghorn B, et al. The expanding phenotype of OFD1 -related disorders: hemizygous loss-of-function variants in three patients with primary ciliary dyskinesia. Mol Genet Genomic Med. 2019;7(9):9. DOI:10.1002/mgg3.911.
- Webb TR, Parfitt DA, Gardner JC, et al. Deep intronic mutation in OFD1, identified by targeted genomic next-generation sequencing, causes a severe form of X-linked retinitis pigmentosa (RP23). Hum Mol Genet. 2012;21(16):3647–3654. DOI:10.1093/hmg/dds194.
- Budny B, Chen W, Omran H, et al. A novel X-linked recessive mental retardation syndrome comprising macrocephaly and ciliary dysfunction is allelic to oral–facial–digital type I syndrome. Hum Genet. 2006;120(2):243–250. DOI:10.1007/s00439-006-0210-5.
- European polycystic kidney dise T. Consortium TEPD The polycystic kidney disease 1 gene encodes a 14 kb transcript and lies within a duplicated region on chromosome 16. Cell. 1994;77(6):881–894.
- Mochizuki T, Wu G, Hayashi T, et al. PKD2, a gene for polycystic kidney disease that encodes an integral membrane protein. Science. 1996;272(5266):1339–1342. DOI:10.1126/science.272.5266.1339.
- Foerster P, Daclin M, Asm S, et al. mTORC1 signaling and primary cilia are required for brain ventricle morphogenesis. Development. 2017;144(2):201–210. DOI:10.1242/dev.138271.
- Yuan S, Li J, Diener DR, et al. Target-of-rapamycin complex 1 (Torc1) signaling modulates cilia size and function through protein synthesis regulation. Proc Natl Acad Sci U S A. 2012;109(6):2021–2026. DOI:10.1073/pnas.1112834109.
- Saxton RA, Sabatini DM. mTOR signaling in growth, metabolism, and disease. Cell. 2017;168(6):960–976.
- McMaster ML, Goldstein AM, Parry DM. Clinical features distinguish childhood chordoma associated with tuberous sclerosis complex (TSC) from chordoma in the general paediatric population. J Med Genet. 2011;48(7):444–449.
- Jacoby M, Cox JJ, Gayral S, et al. INPP5E mutations cause primary cilium signaling defects, ciliary instability and ciliopathies in human and mouse. Nat Genet. 2009;41(9):1027–1031. DOI:10.1038/ng.427.
- Zhang X, Jefferson AB, Auethavekiat V, et al. The protein deficient in Lowe syndrome is a phosphatidylinositol-4,5-bisphosphate 5-phosphatase. Proc Natl Acad Sci U S A. 1995;92(11):4853–4856. DOI:10.1073/pnas.92.11.4853.
- Nickerson ML, Warren MB, Toro JR, et al. Mutations in a novel gene lead to kidney tumors, lung wall defects, and benign tumors of the hair follicle in patients with the birt-hogg-dube syndrome. Cancer Cell. 2002;2(2):157–164. DOI:10.1016/S1535-6108(02)00104-6.