
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
The yeast PROPPIN Atg18 folds as a β-propeller with two binding sites for phosphatidylinositol-3-phosphate (PtdIns3P) and PtdIns(3,5)P2 at its circumference. Membrane insertion of an amphipathic loop of Atg18 leads to membrane tubulation and fission. Atg18 has known functions at the PAS during macroautophagy, but the functional relevance of its endosomal and vacuolar pool is not well understood. Here we show in a proximity-dependent labeling approach and by co-immunoprecipitations that Atg18 interacts with Vps35, a central component of the retromer complex. The binding of Atg18 to Vps35 is competitive with the sorting nexin dimer Vps5 and Vps17. This suggests that Atg18 within the retromer can substitute for both the phosphoinositide binding and the membrane bending capabilities of these sorting nexins. Indeed, we found that Atg18-retromer is required for PtdIns(3,5)P2-dependent vacuolar fragmentation during hyperosmotic stress. The Atg18-retromer is further involved in the normal sorting of the integral membrane protein Atg9. However, PtdIns3P-dependent macroautophagy and the selective cytoplasm-to-vacuole targeting (Cvt) pathway are only partially affected by the Atg18-retromer. We expect that this is due to the plasticity of the different sorting pathways within the endovacuolar system.
Abbreviations: BAR: bin/amphiphysin/Rvs; FOA: 5-fluoroorotic acid; PAS: phagophore assembly site; PROPPIN: beta-propeller that binds phosphoinositides; PtdIns3P: phosphatidylinositol-3-phosphate; PX: phox homology.
Introduction
Macroautophagy, afterward termed autophagy, plays a crucial role in cellular homeostasis and is thus linked to numerous diseases [Citation1]. In S. cerevisiae it starts with the formation of double-membraned autophagosomes [Citation2–4]. In brief, a few vesicles from the trans-Golgi network carrying the membrane protein Atg9 fuse at a scaffolding complex containing the serine/threonine kinase Atg1 to a crescent-shaped phagophore. At the phagophore PtdIns3P is generated, which leads to recruitment of the PtdIns3P-binding proteins Atg18 and Atg21. Atg21 is required for efficient coupling of Atg8 to phosphatidylethanolamine by organizing the Atg8 lipidation machinery at a contact site between the phagophore and the vacuole [Citation5–12]. Lipidated Atg8 interacts with cargo receptors on the inside of the phagophore; on the outside, it is thought to be part of a coat-like structure. For phagophore expansion, Atg18 organizes the Atg2-Atg9 complex at a contact site between the ER and the phagophore. Atg2 delivers lipids from the ER to the growing phagophore, while Atg9 acts as a scramblase and transfers these lipids to the inner membrane leaflet [Citation13]. Eventually, the autophagosome is closed and fuses with the vacuole. Atg9 is recycled back to its trans-Golgi pool either during autophagosome maturation or after fusion with the vacuole [Citation14].
Here, we focus on the molecular functions of Atg18. It folds as a 7 bladed β-propeller, which contains at its circumference two binding sites for PtdIns3P and PtdIns(3,5)P2 [Citation15–19] and is thus termed PROPPIN for β-propeller that binds phosphoinositides. S. cerevisiae contains with Atg21 and Hsv2 two additional homologous members of this protein family. PROPPINS are well conserved and their four mammalian counterparts are termed WIPI and WDR45 proteins [Citation20]. Atg18 not only localizes to the phagophore but also to the vacuole and endosomes [Citation21–23]. At the vacuolar membrane, Atg18 modulates the activity of the PtdIns3P 5-kinase Fab1 and interacts with Vac14, a component of the Fab1-complex [Citation22,Citation24,Citation25]. Atg18 is further required for PtdIns(3,5)P2-dependent vacuolar fragmentation upon hyperosmotic stress and retrograde vacuolar transport of the artificial cargo RS-ALP [Citation21,Citation22,Citation26]. Atg18 contains an amphipathic loop between its two phosphoinositide binding sites. Membrane insertion of this loop leads to membrane tubulation and fission, which is required for its role in vacuole fragmentation [Citation27]. Interestingly, also the mammalian orthologue WIPI1 shares this ability and has been shown to mediate retrograde trafficking from endosomes [Citation28].
To get deeper insight into the molecular function of Atg18, we characterized in a proximity labeling approach the Atg18 microenvironment in living cells and identified the retromer component Vps35 as an Atg18 interactor. Our work shows competitive binding of the SNX-BAR proteins Vps5 and Vps17 and Atg18 with retromer and uncovers the role of Atg18-retromer for vacuole fragmentation upon hyperosmotic stress. Further experiments support a role of Atg18-retromer in Atg9 sorting.
Results
Determination of the Atg18 microenvironment by proximity labeling
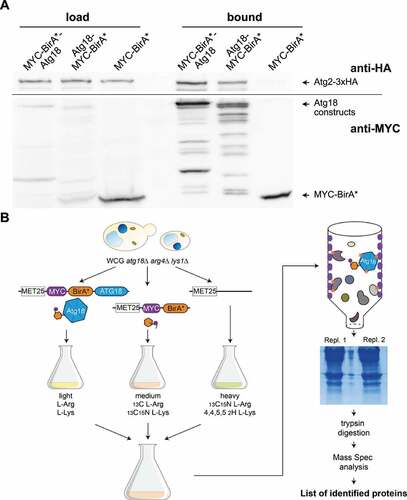
To better understand the molecular function of Atg18, we aimed to identify interacting proteins. To overcome problems with the stability of protein complexes upon cell lysis, we used the proximity-dependent labeling BioID approach [Citation29,Citation30]. This system allows the characterization of protein complexes in living cells. In brief, the E. coli biotin ligase BirA is fused to the protein of interest, which results in the biotinylation of neighboring proteins. After cell lysis and isolation of the biotinylated proteins with a biotin-affinity column, they can be identified by mass spectrometry. To assure promiscuous biotinylation BirA*, a mutated version of the biotin ligase, is used.
We generated both MYC-BirA*-Atg18 and Atg18-MYC-BirA* and found them to be biologically active and expressed (Fig. S1A-C). Next, we checked if the known interaction of Atg18 with Atg2 can be detected with these two constructs. Indeed, Atg2-3xHA was bound to StrepTactin biotin-affinity beads, when either MYC-BirA*-Atg18 or Atg18-MYC-BirA* was expressed, but not with MYC-BirA* alone (). Because MYC-BirA*-Atg18 showed fewer degradation bands, we used this construct for the BioID approach. To quantitatively identify proteins biotinylated by MYC-BirA*-Atg18, but not BirA*, we used a previously established stable isotope labeling by amino acids in cell culture (SILAC) protocol [Citation30]. Here cells expressing MYC-BirA*-Atg18 are grown in medium containing arginine and lysine with light isotopes, while cells expressing MYC-BirA* are grown with amino acids with medium weight isotopes and cells carrying an empty plasmid with heavy isotopes (). The harvested cells were then pooled and biotinylated proteins were isolated by affinity chromatography from their crude extracts. After tryptic digestion the isolated proteins were identified by LC-MS and ranked according to their enrichment compared to the controls. Determination of the known Atg18 interactors Vac14, Fab1 and Atg2 together with Atg18 self biotinylation confirmed the validity of our approach (). Interestingly, we further identified with Vps35 and Snx3 two components of the retromer complex.
Table 1. Identification of Atg18 interactors with the BioID approach.
Figure 1. Characterization of the Atg18 microenvironment. (A) A small-scale proximity-dependent labeling BioID approach was performed to test the functionality of the biotin ligase (BirA*) fusion constructs of Atg18 with the known interactor Atg2. Cells expressing the BirA* constructs together with Atg2-3xHA were grown in selection medium supplemented with biotin, harvested and lysed. Biotinylated proteins were purified with Strep-Tactin columns and visualized in Western blots with antibodies against HA and MYC. (B) Workflow for the BioID assay. WCG atg18∆ arg4∆ lys1∆ strains expressing under the control of a MET17 promotor either MYC-BirA*-Atg18, MYC-BirA* or an empty plasmid were grown in selection medium supplemented with methionine, biotin and isotope labeled arginine and lysine. Pooled cultures were then lysed and biotinylated proteins purified using Strep-Tactin columns. After SDS-PAGE the proteins were digested with trypsin and analyzed using mass spectrometry.
Atg18 coprecipitates with the retromer component Vps35, but not with Snx3
Next, we wanted to confirm the putative interaction of Atg18 with Vps35 and Snx3 in co-immunoprecipitation experiments. To avoid artifacts caused by non-endogenous expression levels, Vps35 was chromosomally tagged at its carboxy-terminus with 6xHA and Atg18-GFP was expressed from its endogenous promotor. Indeed, Vps35-6xHA precipitated with Atg18-GFP, but not with GFP alone (), as a control, the known Atg18 interactor Atg2 was included. Since we could not detect an interaction between Atg18 and Snx3 under these conditions, GFP-Atg18 was further expressed with the MET17 promotor in the presence of 0.3 µM methionine. Again, the interaction of Atg18 with Vps35 was clearly detectable (). Furthermore, the interaction of GFP-Atg18 with Vac14-6xHA could be confirmed, which was previously only detectable using the yeast two-hybrid system [Citation25]. However, again no interaction between GFP-Atg18 and Snx3-6xHA could be seen under these conditions. Also, after starvation of the cells for 2 h in a nitrogen-free SD-N medium only interaction of Atg18 with Vps35, but not with Snx3 was detectable. Together, this suggests that Snx3 is not an interactor of Atg18, but it might be present in its vicinity.
Figure 2. Vps35 interacts with Atg18. (A) Vps35 co-precipitates with endogenous levels of Atg18. Immunoprecipitations were performed with atg18∆ cells expressing Atg2-3xHA or Vps35-6xHA together with GFP or Atg18-GFP. Atg2 was included as a known Atg18 interactor and could be detected in the bound fraction of Atg18-GFP, but not GFP alone. (B) GFP-Atg18 overexpression increases co-immunoprecipitation. GFP-Atg18 was expressed from a MET17 promotor in the presence of Vps35-6xHA, Vac14-6xHA and Snx3-6xHA, respectively. This increased the amount of Vps35-6xHA bound to GFP-beads, and also allowed detection of Vac14 in the bound fraction of GFP-Atg18. No interaction between Snx3 and Atg18 could be seen. (C) Interaction with Vps35-6xHA is reduced for GFP-Atg18FTTG, which due to defective phosphoinositide binding localizes to the cytosol. (D) Quantification of the co-immunoprecipitation of GFP-Atg18 and GFP-Atg18FTTG with Vps35-6xHA. The ratios were normalized to the ratio of GFP-Atg18. (E, F) Immunoprecipitation of Vps35 with Atg18 is reduced in the absence of Vps30. A Co-IP was performed with either wildtype or vps30∆ cells expressing Vps35-6xHA and GFP-Atg18 or GFP as a control. At least three independent experiments were quantified and the ratio of HA signal to GFP signal was normalized to the wildtype. Statistical relevance was determined using an unpaired two-tailed t-test, error bars indicate SEM, asterisks indicate p-values. (G) Vps35 colocalizes with Atg18. Vps35 was chromosomally tagged with mCherry and its localization was analyzed compared to endogenously expressed Atg18-GFP. White arrows indicate puncta positive for both Atg18 and Vps35. Scale bar: 5 µm.
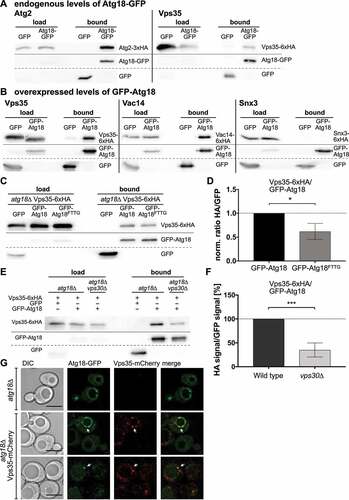
Membrane association of Atg18 depends on the binding to two PtdIns3P molecules via a conserved FRRG motif, accordingly a mutated Atg18-FTTG variant localizes to the cytosol. Co-immunoprecipitation of GFP-Atg18 with Vps35-6xHA was significantly reduced with Atg18-FTTG but still preserved, suggesting that the interaction is favored by membrane association, but can also occur in the cytosol (). The reduced but still clearly detectable coimmunoprecipitation of GFP-Atg18 with Vps35-HA in vps30∆ cells, lacking the autophagy specific PtdIns 3-kinase complex, strengthen this view (). In line with an interaction of Atg18 and Vps35, fluorescence microscopy showed that 30% of the Atg18-GFP puncta colocalized with chromosomally expressed Vps35-mcherry ().
Cross talk between Atg18-retromer and Vps5-Vps17-retromer
In S. cerevisiae the retromer complex recycles proteins and lipids from the endosome to the Golgi. It contains the cargo selection complex or simply retromer, which consists of Vps26, Vps29 and Vps35 and forms arch-like structures () [Citation31,Citation32]. Retromer further interacts with a dimeric complex of SNX-BAR sorting nexins such as Vps5 and Vps17. Vps5 and Vps17 bind via their PX domain to PtdIns3P-positive membranes and induce membrane bending with their BAR domain. This leads to the assembly of a spiral coat and is thought to induce membrane tubulation and the formation of endosomal transport carriers.
Figure 3. Relationship between Atg18 and components of the retromer complex (A) Model of the retromer complex containing Vps35, Vps26 and Vps29 together with the SNX-BAR proteins Vps5 and Vps17. (B) Co-precipitation of Vps35-6xHA with GFP-Atg18 was increased in the absence of either Vps5 or Vps17. Vps35-6xHA was chromosomally expressed, while GFP-Atg18 was under the control of a MET17 promotor. (C) The amount of Vps35-6xHA bound to GFP-Atg18 is decreased in a vps26∆ strain and no binding could be detected in a vps29∆ strain. (D) Quantification of at least three independent co-immunoprecipitation experiments. The ratio of HA signal to GFP signal in the bound fraction was normalized to the wildtype. Statistical relevance was determined using an unpaired two-tailed t-test, error bars indicate SEM, asterisks indicate p-values. (E,F) Vps26 co-precipitates with Atg18 in the presence of Vps35, while Vps29 does not. Either Vps26 or Vps29 were chromosomally tagged with 6xHA and expressed together with GFP-Atg18 under a MET17 promotor control. (G,H) Vps5 competes with Atg18 for binding to Vps35 during nitrogen starvation. Vps35-6xHA was expressed chromosomally together with either GFP-Atg18 (G) or Vps5-GFP (H) under the control of a MET17 promotor. Co-IPs were performed before and after 2 h of nitrogen starvation. Either Vps5 (G) or Atg18 (H) were deleted (-) or constitutively overexpressed under the control of a GPD1 promotor (+++). (I,J) Quantification of at least three independent co-immunoprecipitation experiments. The ratio of HA signal to GFP signal in the bound fraction was normalized to the 0 h sample of the wildtype. Statistical relevance was determined using an unpaired two-tailed t-test, error bars indicate SEM, asterisks indicate p-values.
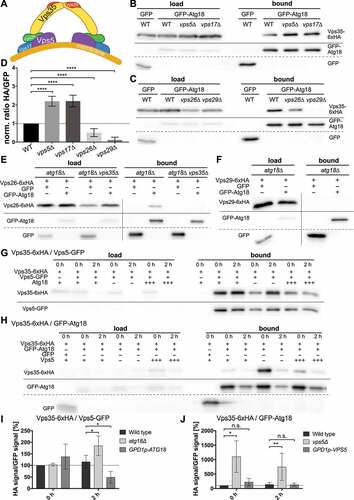
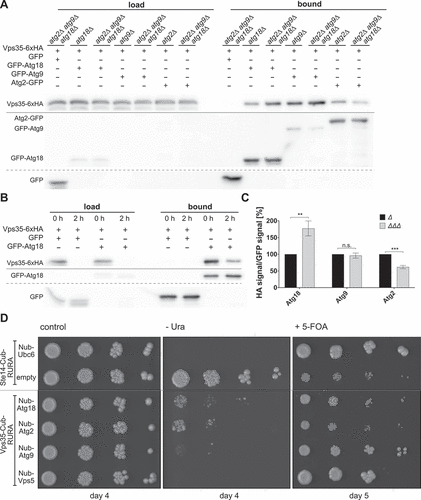
Remarkably, the amount of Vps35-6xHA co-immunoprecipitating with GFP-Atg18 was more than two-fold higher in the absence of either Vps5 or Vps17 (), suggesting competitive binding of Atg18 and the sorting nexins to retromer. Indeed, overexpression of Atg18 from the strong GPD1-promotor significantly lowered the coimmunoprecipitation of Vps35-HA and Vps5-GFP after starvation for 2 h (). However, overexpression of Vps5 had no further inhibiting effect on the interaction of Vps35 with Atg18, suggesting that normal Vps5 levels are sufficient to balance the formation of the different retromer complexes ().
We further analyzed whether or not the interaction of Atg18 with Vps35 depends on the presence of the other retromer subunits. Well in line with the important role of Vps29 in retromer assembly [Citation32], in the absence of Vps29 almost no Vps35-6xHA coprecipitated with GFP-Atg18, while approximately half of it was still bound in theabsence of Vps26 (). Further co-immunoprecipitation experiments showed that Vps26-6xHA is bound by GFP-Atg18, but only in the presence of Vps35 (). For Vps29-6xHA we could not detect an interaction with GFP-Atg18 (). Taken together, our findings suggest the formation of an Atg18-retromer complex, and competitive binding of retromer by either Atg18 or Vps5 and Vps17.
Hyperosmotic vacuole fragmentation depends on Atg18-retromer
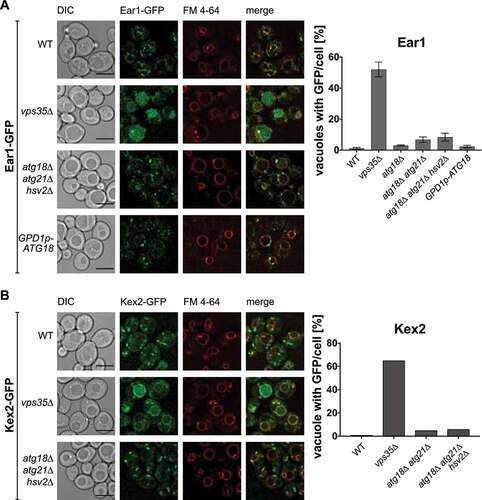
One of the best characterized retromer functions is the intracellular trafficking of endosomal cargo proteins such as Kex2 and Ear1 [Citation31,Citation33]. Retromer defects typically lead to mislocalization of the cargos to the vacuole. Indeed, compared to wild-type cells vps35∆ cells showed drastically increased vacuolar localization of Ear1-GFP and Kex2-GFP, which was not the case for atg18∆ atg21∆ hsv2∆ cells or cells overexpressing Atg18 (). We used cells lacking all three yeast PROPPINs to exclude the possibility that lack of Atg18 might be masked by one of the other homologues. We conclude that Atg18-retromer might have functions distinct from Vps5/Vps17-retromer and Snx3-retromer.
Figure 4. Sorting of Ear1 and Kex2 appears unaffected by Atg18, Atg21 and Hsv2. Ear1 (A) and Kex2 (B) were C-terminally tagged with GFP and expressed from the chromosome in either wildtype, the indicated deletion strains or, in case of Ear1, with overexpressed Atg18. FM 4–64 was used to stain the vacuolar membrane. Cells with increased GFP signal inside the vacuole were counted and the ratio of cells with stained vacuole to total amount of cells was calculated. Scale bar: 5 µm.
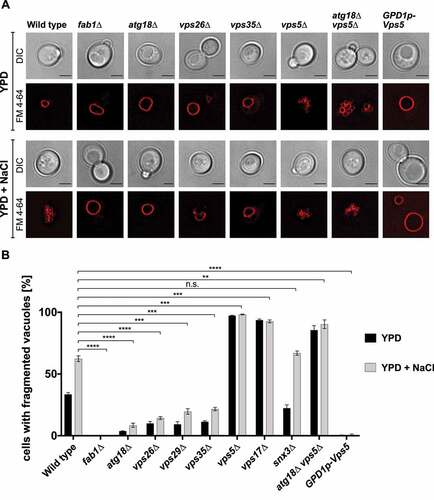
We were further interested, which of the previously described Atg18 functions depends on retromer. The role of Atg18 in regulating vacuolar morphology and especially the fragmentation of vacuoles upon hyperosmotic stress is well established [Citation21,Citation22,Citation24,Citation26,Citation27,Citation34]. We thus labeled the vacuoles with the fluorescent dye FM 4–64 and counted in the presence and absence of 0.4 M NaCl the percentage of cells exhibiting fragmented vacuoles. In agreement with published data, atg18∆ showed a severe defect in vacuole fragmentation compared to wild-type cells (). Remarkably, the retromer defects in vps26∆, vps29∆ and vps35∆ cells caused comparable defects in vacuolar fragmentation, suggesting a role of Atg18-retromer in this process. On the other side, deletion of either VPS5 or VPS17 caused hyper-fragmentation of the vacuoles upon hyperosmotic stress, while overexpression of Vps5 with the strong GPD1-promotor blocked vacuole fragmentation almost completely (). This observation strengthens the crosstalk between the Atg18- and the Vps5/Vps17-retromer shown in . As expected, snx3∆ cells showed normal vacuole fragmentation. vps5∆ atg18∆ double-deleted cells were still highly fragmented, which might be due to compensatory effects of the other PROPPINs or due to binding of the free CSC to Ypt7, reducing its fusion activity. While Vps5-GFP under growing and starvation conditions showed a punctate localization (Fig. S2), Atg18-GFP showed an increased punctate localization in growing and starved vps5∆ cells.
Figure 5. Atg18-retromer affects hyperosmotic vacuolar fragmentation. (A) Cells deleted for retromer subunits and fab1∆ cells were stained with FM 4–64 and either incubated in YPD medium or shifted to hyperosmotic medium containing 0.4 M NaCl. After 1 h vacuolar fragmentation was inspected in fluorescence microscopy. Scale bar: 5 µm. (B) The percentage of cells containing fragmented vacuoles were determined in at least three independent experiments. Statistical relevance was determined using an unpaired two-tailed t-test, error bars indicate SEM, asterisks indicate p-values.
Atg9 interacts with Vps35 and is missorted in starved vps35∆ cells
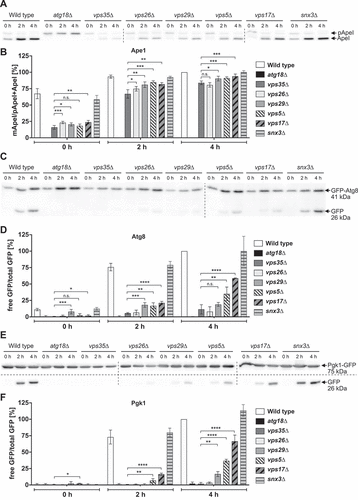
Atg18 further acts during autophagy by organizing the complex of Atg2 with Atg9, which delivers and distributes lipids to the growing phagophore [Citation13]. In addition, Atg18 is required for recycling of Atg9 to a post-Golgi pool [Citation23]. We wondered if Atg18-retromer might be involved in these autophagy related Atg18 functions. To this end, we analyzed the autophagic activity in vps35∆ cells. Maturation of prApe1 was only slightly but significantly reduced in both growing and nitrogen-starved vps35∆ cells (). To measure the autophagy rate in immunoblots, we took advantage of the accumulation of rather proteolysis resistant GFP inside the vacuole [Citation35]. Degradation of GFP-Atg8 was reduced to about 20% of the wild-type level after 4 hours in nitrogen-free SD-N medium (), while the unselective autophagy marker Pgk1-GFP [Citation36] was degraded with about 70% of the normal rate (). As expected, vps5∆ and vps17∆ cells showed significantly higher autophagy rates compared to vps35∆ cells (). Together, our data indicate that autophagy in SD-N medium is not blocked in the absence of Vps35, but significantly reduced. This might either be due to an indirect role of Vps35 in autophagy or the plasticity of the endovacuolar system, where alternative transport routes can probably compensate for defects of others.
Figure 6. Effect of retromer on autophagic activity. The level of prApe1 maturation was determined in immunoblots (A) and the percentage of mApe1 was calculated (B). Autophagic breakdown of GFP-Atg8 (C, D) as well as a Pgk1-GFP assay to determine nonselective autophagy (E,F) was analyzed in immunoblots and the amount of rather proteolysis stable GFP was quantified. Cells were nitrogen starved in SD-N and harvested for analysis. The prApeI maturation rate was determined by dividing the amount of mature ApeI to the total amount of Ape1, while the ratio of free GFP to total GFP was calculated for both the GFP-Atg8 and Pgk1-GFP assay. At least three independent experiments were quantified. Statistical relevance was determined using an unpaired two-tailed t-test, error bars indicate SEM, asterisks indicate p-values.
We thus extended our analyses of the Atg18 and Vps35 interactome onto Atg9 and Atg2. Co-immunoprecipitation of Vps35 with Atg18 was effective in cells lacking both Atg9 and Atg2 (), accordingly the absence of Atg2 alone or starvation for 2 hours in nitrogen-free medium did not abrogate the interaction (). More importantly, co-immunoprecipitation also showed an interaction between Vps35 and Atg9 in wild-type cells. In the absence of both Atg18 and Atg2 the interaction was clearly detectable (). In line with the non-essential role of Atg2 for the interaction of Vps35 with both Atg9 and Atg18, only a weak interaction of Vps35 with Atg2 was detected in both wild-type and atg18∆ cells (). Additionally, we used the split-ubiquitin system, which is similar to the 2-hybrid system, but better suited to detect interactions of membrane associated proteins [Citation37]. We generated Vps35-Cub, tagged at its C-terminus with the carboxyterminal domain of ubiquitin. As a positive control we used Nub-Vps5, carrying the amino-terminal domain of ubiquitin at its amino-terminus. Upon interaction both ubiquitin domains come together and ubiquitin is removed by deubiquitinating enzymes, releasing a destabilized variant of the Ura3 protein, which is then rapidly degraded by the proteasome. Thus, interaction leads to uracil-auxotroph cells unable to grow without uracil in the medium. Additionally, growth on 5-fluoroorotic acid (5-FOA) is analyzed, here cells auxotroph for uracil can grow. We detected the known strong interaction of Vps35 with Vps5 (). Well in line with our co-immunoprecipitations, also Atg9 showed a strong interaction. Atg18 and Atg2 showed a weaker, but clearly detectable interaction. It should be mentioned that lack of interaction in the split-ubiquitin system might be due to steric reasons, since both ubiquitin domains must come in close contact. Thus, negative results or weak interactions need verification by coimmunoprecipitations.
Figure 7. Vps35 interacts with Atg9 and Atg2 independent of Atg18. (A) Vps35 co-precipitates with Atg18, Atg2 and Atg9 independent of each other. Vps35-6xHA was expressed chromosomally together with either GFP-Atg18, Atg2-GFP or GFP-Atg9 in the presence or absence of chromosomally expressed Atg2, Atg9 and Atg18. (B) Vps35 co-precipitates with Atg18 under starvation conditions. Cells expressing both Vps35-6xHA and GFP-Atg18 were incubated for 2 h in nitrogen free medium, before a Co-IP was performed. (C) Quantification to (A) of at least three independent co-immunoprecipitation experiments. The ratio of HA signal to GFP signal in the bound fraction was normalized to the wildtype. Statistical relevance was determined using an unpaired two-tailed t-test, error bars indicate SEM, asterisks indicate p-values. (D) Split-Ubiquitin assay: tenfold serial dilutions of cells expressing Cub (bait) and Nub (prey) constructs were grown on plates with CM -His -Trp (growth control), CM -His -Trp -Ura (growth indicates no interaction) and CM -His -Trp +5-FOA (growth indicates interaction). Ste14-Cub/Ubc6-Nub: positive control, Ste14-Cub/empty: negative control.
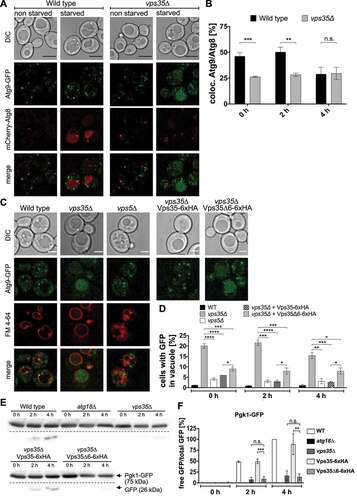
Together our data would fit with the existence of an Atg18-retromer complex containing Atg9, which might be involved in the recycling of Atg9 to its trans-Golgi pool. We therefore next analyzed in fluorescence microscopy the localization of Atg9-GFP and mCherry-Atg8 in vps35∆ cells. Compared to wild-type cells vps35∆ cells showed after 0 and 2 hours in a nitrogen-free medium a reduced number of Atg8 puncta, which were also positive for Atg9 (). In contrast to wild-type cells, vps35∆ cells showed the appearance of GFP fluorescence in their vacuoles, with a maximum at 2 hours after shifting to SD-N medium, while in vps5∆ cells only very few vacuoles showed GFP fluorescence (). Since Atg9-GFP is a membrane protein with its carboxy terminus exposed to the cytosol this indicates its disturbed sorting in vps35∆ cells, most likely leading to entry into the MVB-pathway and its release into the vacuole.
Figure 8. Atg9 is mislocalized in vps35∆ cells. (A) Chromosomally tagged Atg9-GFP was expressed together with mCherry-Atg8 under the control of an endogenous promotor in either wild-type or vps35∆ cells. Cells were grown to an OD600 of ~2 and transferred to SD-N to induce autophagy. Black scale bar: 5 µm. (B) Puncta positive for both Atg9-GFP and mCherry-Atg8 were counted and the percentage of colocalization in relation to the total number of Atg8 puncta was calculated in at least three independent experiments. (C) Atg9-GFP mislocalizes to the vacuole in the absence of functional Vps35. Wild-type, vps35∆ or vps5∆ cells expressing Atg9-GFP were nitrogen starved and stained with FM 4–64 to mark the vacuolar membrane. Deletion of repeat6 in Vps35 affects Atg9 localization. White scale bar: 2.5 µm. (D) Cells with increased GFP signal inside the vacuole were counted and the ratio of cells with stained vacuole to the total number of cells was determined. (E, F) Effect of Vps35∆6 on autophagic activity. Autophagic degradation of Pgk1-GFP was analyzed after 0, 2, and 4 h of nitrogen starvation, and the ratio of free GFP to total GFP was calculated. Statistical relevance was determined using an unpaired two-tailed t-test, error bars indicate SEM, asterisks indicate p-values.
We generated a plasmid expressing the vps35∆repeat6 mutant, which is defective in Ypt7 binding [Citation38–40] and measured the mislocalization of Atg9-GFP to the vacuole lumen in growing and starved cells. As shown in expression of vps35∆repeat6 reduced the Atg9 vacuolar mislocalization rate to about 2/3 of that seen in vps35∆ cells, but not to the level upon expression of Vps35. Interestingly, the autophagy rate measured by breakdown of Pgk1-GFP () was comparable in vps35∆ cells and in cells expressing Vps35∆repeat6, supporting a crucial role of Vps35 binding to Ypt7 for autophagy. In sum, the data support the idea that defects in Atg9 cycling lead to its uptake at endosomes into MVB-vesicles, which cause its appearance in the vacuole.
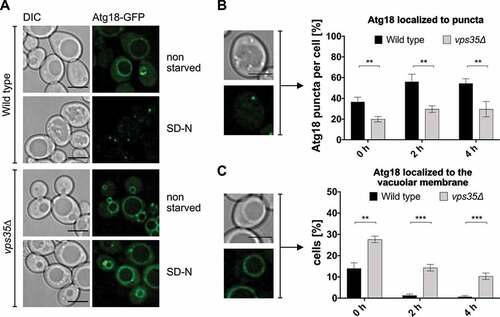
Fluorescence microscopy showed that compared to wild-type cells Atg18-GFP exhibited in vps35∆ cells a reduced number of puncta per cell, but an increased vacuolar membrane localization (). This altered intracellular localization further supports an additional role of Atg18-retromer in protein trafficking.
Figure 9. Atg18 accumulates at the vacuolar membrane in the absence of Vps35. (A) Wild-type or vps35∆ cells expressing Atg18-GFP under the control of an endogenous promotor were grown to an OD600 of 2–3 and transferred to nitrogen free SD-N. Localization of Atg18-GFP was then analyzed using fluorescence microscopy. Scale bar: 5 µm. (B) Atg18-GFP puncta in the cytosol were counted and the ratio of puncta per total number of cells was determined. (C) Cells with Atg18-GFP localized at the vacuolar membrane were counted and divided by the total number of cells to calculate the percentage of cells with GFP at the vacuole membrane. (B, C) Three independent experiments were analyzed for quantification. Statistical relevance was determined using an unpaired two-tailed t-test, error bars indicate SEM, asterisks indicate p-values. Scale bar: 4 µm.
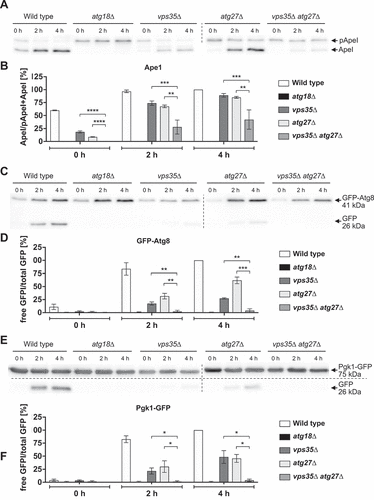
In addition, we generated an atg27∆ vps35∆ double-deleted strain and measured the autophagy rate using breakdown of Pgk1-GFP. vps35∆ and atg27∆ cells both showed about half of the wild-type autophagy rate, while autophagy in the atg27 vps35 double-deleted cells was almost blocked (). Also, GFP-Atg8 breakdown () and prApe1 maturation () showed are clear additive effect. This shows that alternate routes for Atg9 cycling exist, which underlines the plasticity of the endovacuolar sorting system.
Figure 10. Autophagic activity vps35∆ atg27∆ cells. The level of prApe1 maturation was determined in immunoblots (A) and the percentage of mApe1 was calculated (B). Autophagic breakdown of GFP-Atg8 (C, D) as well as a Pgk1-GFP assay to determine nonselective autophagy (E,F) was analyzed in immunoblots and the amount of rather proteolysis stable GFP was quantified. Cells were nitrogen starved in SD-N and harvested for analysis. The prApeI maturation rate was determined by dividing the amount of mature ApeI to the total amount of Ape1, while the ratio of free GFP to total GFP was calculated for both the GFP-Atg8 and Pgk1-GFP assay. At least two independent experiments were quantified. Statistical relevance was determined using an unpaired two-tailed t-test, error bars indicate SEM, asterisks indicate p-values.
Discussion
Our proximity-dependent labeling approach for Atg18 identified besides the known interactors Vac14, Fab1 and Atg2 the retromer subunit Vps35 ( and ). Vps35 assembles together with Vps26 and Vps29 into the arch-like cargo selective retromer complex often simply called retromer (). At endosomes retromer further associates with a dimeric complex of the sorting nexins Vps5 and Vps17 into spiral-like structures. These sorting nexins bind via their PX domains to PtdIns3P and induce with their BAR domains membrane tubulation and finally scission of endosomal transport carriers. Our co-immunoprecipitations showed that the interaction of Atg18 with Vps35 requires Vps29 suggesting that Atg18 interacts with the assembled arch-like retromer (). Indeed, we observed binding of Atg18 to Vps26 only in the presence of Vps35. Based on the significantly strengthened interaction of Atg18 with Vps35 in the absence of Vps5 or Vps17, we expect that retromer can either associate with the dimeric sorting nexins or with Atg18. This is confirmed by the almost complete block of vacuole fragmentation when Vps5 is overexpressed () and the significantly reduced interaction of Vps35 with Vps5 after 2 h starvation in cells overexpressing Atg18 (). The typical cargos of Vps5/Vps17-retromer, such as Ear1 and Kex2, are not mislocalized in the absence of Atg18 (); underscoring the different roles of these retromer complexes. Furthermore, Atg18 shows features allowing it to functionally substitute for the sorting nexins. As a PROPPIN Atg18 does not contain a PX domain but two binding sites for PtdIns3P and PtdIns(3,5)P2. Instead of the BAR domain the loop CD in blade 6 of Atg18 can fold as an amphipathic helix, which penetrates into the membrane and thus generates membrane bending. It has been shown that Atg18 and especially its loop 6 CD plays a crucial role in vacuolar fragmentation upon hyperosmotic stress [Citation21,Citation26,Citation27]. Since Vps26, Vps29 and Vps35 are also required for efficient vacuolar fragmentation during salt stress while deletion of VPS5 and VPS17 resulted in hyper-fragmentation (), we expect that Atg18-retromer acts during vacuolar fragmentation. It has been proposed that the absence of Vps5 or Vps17 leads to the increased interaction of the Rab-GTPase Ypt7 with retromer and that Ypt7 is thus not available for vacuole fusion [Citation38–40]. Indeed, a vps35∆repeat6 mutant unable to bind Ypt7 was almost blocked in autophagy of Pgk1-GFP () and reduced Atg9 mislocalization to the vacuole lumen, but not the level seen in cells expressing Vps35 ().
Our proximity-dependent labeling approach further identified Snx3. However, in coimmunoprecipitations we could not verify an interaction of Atg18 with Snx3, even after starvation and Atg18 overexpression. This might be due to the experimental conditions used; however, we can also envision an alternate scenario. The relation between Snx3-retromer and Vps5-Vps17-retromer is not fully understood. Since Snx3 only contains a PX-domain, it has been speculated to act preferentially in cargo recognition [Citation31,Citation33]. One idea is that one complex might act during cargo recognition, while tubulation and scission of endosomal transport carriers are mainly mediated by the other. For further studies, it should also be taken into account that some Atg18-retromer might act in the proximity of Snx3-retromer.
The membrane deformation and scission activity of Atg18 and mammalian WIPI1 depends on insertion of the amphipathic helix formed by loop 6 CD into the membrane [Citation27,Citation28]. Mutations abrogating the formation of an amphipathic helix abolished this function, without significantly affecting the autophagic rate. This suggests that autophagy does not require the fission activity of these proteins. For WIPI1 tubulation at early endosomes was dependent on PtdIns3P, while fission required PtdIns(3,5)P2. Similarly, in S. cerevisiae autophagy requires PtdIns3P, while vacuolar fragmentation upon hyperosmotic stress depends on the PtdIns3P 5-kinase Fab1.
We were thus further interested if Atg18-retromer plays a role in autophagy and especially in the recycling of Atg9. Atg18 itself is required for autophagosome elongation and in its absence Atg9 is restricted to the phagophore. We therefore focused on the central retromer component Vps35. In vps35∆ cells, selective autophagy followed by maturation of prApe1 was slightly but significantly reduced. Unselective macroautophagy followed by Pgk1-GFP breakdown was reduced for about 30% after 4 hours in a nitrogen-free medium and GFP-Atg8 breakdown was reduced to ~20% of the wild type level (). In sum, retromer is required for efficient autophagy, but not essential. Remarkably, the split-ubiquitin system indicated an interaction of Vps35 with Atg9 (). Co-immunoprecipitation experiments confirmed the interaction of Vps35 with Atg9, which was still present in atg2∆ atg18∆ double-deleted cells (). Also, the interaction of Atg18 with Vps35 was independent of the presence of Atg9 and Atg2. This would fit with Atg9 being a cargo of Atg18-retromer. Indeed, fewer Atg8-puncta were also positive for Atg9 and most interestingly after 2 h of nitrogen-starvation, Atg9-GFP was missorted to the vacuolar lumen (). In addition, in vps35∆ cells less Atg18-puncta, but increased localization to the vacuolar membrane was detectable (). We cannot exclude that lack of Vps35 causes general imbalances within the endovacuolar system, which indirectly affect autophagy and Atg9 sorting. Based on the observed interaction of Atg9 with Vps35, we however favor another explanation. Retrograde transport from the vacuole to endosomes and the Golgi is mediated by several pathways, which can act in parallel and might partially compensate for one another.
Snx24 (Atg24), another SNX-BAR protein, acts in two dimeric protein complexes either together with Snx41 or Atg20 (Snx42) [Citation41,Citation42]. Atg27, which is involved in Atg9 trafficking is sorted Snx4-dependent from the vacuole to endosomes and retromer-dependent from endosomes to the Golgi and deletion of ATG27 leads to missorting of Atg9 to the vacuole lumen [Citation33,Citation43,Citation44]. In yeast, Atg9 sorting appears to occur by Snx4 and in a redundant pathway by retromer [Citation45,Citation46]. In higher eukaryotes, ATG9A traffic from endolysosomes to early endosomes requires SNX4, while its transit from early endosomes to the Golgi is mediated by VPS35. Reminiscent to the partial autophagy defect we found in vps35∆ cells, silencing of VPS35 in higher cells only reduced the starvation-induced protein degradation by ~40% [Citation47]. The importance of VPS35 for ATG9A trafficking is underscored by sorting defects observed with VPS35D620N. This mutated variant was identified in patients with Parkinson disease [Citation48]. Together, we thus expect that Atg9 recycling in the absence of Atg18-retromer might be bypassed to some extend by other pathways. Indeed, vps35∆ and atg27∆ cells both showed about half of the wild-type autophagy rate measured with Pgk1-GFP, while autophagy in the atg27∆ vps35∆ double-deleted cells was almost blocked ().
At the moment it is not understood how the assembly of Atg18 into different protein complexes is regulated. Hints for such a regulatory mechanism might come from a study reporting the regulation of membrane association and PtdIns(3,5)P2-binding by phosphorylation of the loop 6 CD and blade 7 [Citation49]. Further studies will be necessary to shed light on those mechanisms.
Materials and methods
Strains, media and growth conditions
Yeast strains listed in are derived from the WT strain WCG4 MATα his3-11,15 leu2-3,112 ura3 [Citation50] and BY MATa; his3∆1, leu2∆0, met15∆0, ura3∆0. Deletions and insertions were generated using the method described by [Citation51,Citation52].
Table 2. Strains used.
Yeast strains were cultivated at 30°C and 220 rpm in yeast extract peptone dextrose medium (YPD, 1% yeast extract, 2% peptone, 2% glucose, pH 5.5) or synthetic complete medium (CM, 0.67% yeast nitrogen base w/o amino acids, 2% glucose, pH 5.6, supplemented with 0.0117% of the appropriate amino acids for auxotrophic selection). Autophagy was induced by nitrogen starvation: yeast cells were incubated in synthetic defined medium w/o nitrogen (SD-N, 0.17% yeast nitrogen base w/o amino acids and ammonium sulfate, 2% glucose).
Chromosomal modifications of the yeast genome to introduce C- or N-terminal tags, deletions or alternative promotors were performed as described previously [Citation51–54].
Plasmids
Plasmids used in this study are listed in . For the construction of pUG36-MYC-BirA*-MCS the insert MYC-BirA* was amplified lacking a stop codon and bracketed by XbaI restriction sites. Amplification products as well as the backbone pUG36 were digested with XbaI and ligated, replacing the GFP of the backbone with MYC-BirA*. Atg18 was inserted into the MCS by digesting both vector and insert with XhoI and SpeI.
Table 3. Plasmids used.
GFP tagged Atg18 and Atg2 were generated by copying the respective gene with its endogenous promotor from chromosomal DNA. Restriction sites EcoRI/SacI for Atg18 and SalI/SpeI for Atg2 were used for insertion into the backbone pUG23 (J. H. Hegemann, Heinrich-Heine-University, Duesseldorf, Germany).
For Vps35∆repeat6 Vps35 was chromosomally tagged with 6xHA and then copied including its promotor with oligos containing restriction sites for SacI and SmaI. The PCR product was then inserted into the backbone pUG36 replacing GFP and the MET17 promotor. The NEBuilder® HiFi DNA Assembly Cloning Kit (NEB, E5520S) was used to delete repeat 6 of Vps35: oligos sharing complementary ends were designed to bind to both sites of the deletion sites and copy the whole plasmid. The overlapping ends were then annealed to form a plasmid coding for Vps35∆repeat6-6xHA.
To generate Vps35-Cub-RURA3 Vps35 was amplified from the chromosome with oligos containing the restriction sites for EcoRI and SalI and ligated in pRS313-MET17-Cub-RURA3.
Western blot analysis
Two OD600 units of yeast cells were pelleted (3,000 x g, 5 min) and incubated for 10 min on ice in 1 ml alkaline lysis buffer (0.28 M NaOH, 1.125% [v:v] β-mercaptoethanol). Proteins were precipitated with trichloroacetic acid (TCA) in a final concentration (f.c.) of 7.5% (v:v), pelleted (18,000 x g, 10 min, 4°C), washed twice with ice cold acetone, dried and resuspended in 2x Lämmli buffer (117 mM Tris, pH 8, 3.4% [w:v] SDS, 12% [w:v] glycerol, 0.004% [w:v] bromophenol blue, 0.016% [w:v] β-mercaptoethanol). The proteins were then analyzed using SDS-PAGE and western blotting, with rabbit anti-ApeI antibodies [Citation57] in a dilution of 1:5,000 and mouse anti-GFP monoclonal antibodies (Roche Diagnostics, 11814460001) in a dilution of 1:10,000. Goat anti-rabbit (ThermoFisher Scientific, G-21234) and goat anti-mouse monoclonal antibodies conjugated to horseradish peroxidase (Dianova, 115–035-166) were used as second antibodies, respectively, and detected with either PierceTM ECL Plus Western Blotting Substrate (ThermoFisher Scientific, 32132) or AmershamTM ECL Western-Blotting Detection Reagents (GE Healthcare, RPN2106).
Biotin ligation (BioID) approach
Strains expressing either MYC-BirA*-Atg18, MYC-BirA* or an empty plasmid were cultivated individually in CM supplemented with 10 µM D-biotin (Sigma-Aldrich, 2031–1 GM) and the appropriate amino acids including 50 mg/l of stable isotope labeled amino acids (Silantes GmbH, 13C6-L-arginine HCl [201203902], 13C6 15N4-L-arginine HCl [201603902], 4,4,5,5-D4-L-lysine HCl [211103913], 13C6-L-lysine HCl [211203902]). Cells were grown overnight to an OD600 of approximately 3 and 200 OD600 units were harvested (720 x g, 5 min, 4°C) and washed twice in 15 ml ice cold 10 µM HEPES, pH 7.9. The three cell suspensions were pooled in a 1:1:1 ratio according to the optical densities of the cultures, centrifuged (720 x g, 10 min, 4°C) and resuspended in lysis buffer (10 mM HEPES, pH 7.9, 10 mM KCl, 1.5 mM MgCl2, 1 mM PMSF [Roth, 6367.1], 1x 25x c0mplete, EDTA free [Roche Diagnostics, 34044100], 0.5 mM DTT). Glass beads were added and cells were vortexed for 30 min at 4°C to break the cell wall. SDS was added to a final concentration of 4% (w:v) and the samples were incubated for 10 min at 65°C. Glass beads and cell debris were removed by centrifugation (3,000 x g, 5 min, room temperature). Biotinylated proteins were isolated with Strep-Tactin Sepharose Columns (iba Lifesciences, 2–1202-051) according to the protocol established by Opitz et al. [Citation30] and modified by Schmitt & Valerius [Citation58]. Proteins were concentrated using TCA precipitation (f.c. of 10% [w:v] TCA) and resuspended in 2x Lämmli buffer (117 mM Tris, pH 8, 3.4% [w:v] SDS, 12% [w:v] glycerol, 0.004% [w:v] bromophenol blue, 0.016% [w:v] β-mercaptoethanol). Biotinylated proteins were then separated by size using SDS-PAGE, stained with Coomassie Brilliant Blue [Citation59,Citation60] . The gel was divided into ten sections and proteins were digested in-gel with trypsin. Samples were then purified using C18 (3 M) stop and go extraction (stage) tips and dried before subjecting to LC-MS analysis [Citation58,Citation61].
A small-scale approach was performed to test the function of the fusion proteins with known interaction partners. For this experiment non labeled amino acids were added to the medium and isolated biotinylated proteins were analyzed with SDS-PAGE and western blotting. GFP and HA were detected with mouse anti-GFP antibodies (Roche Diagnostics, 1181460001) diluted 1:1000 and mouse anti-HA monoclonal antibodies (Santa Cruz Biotechnology, sc-7392) diluted 1:10,000, respectively. Second antibodies were horseradish peroxidase linked goat anti-mouse antibodies (Dianova, 112–035-003) detected with PierceTM ECL Plus Western Blotting Substrate (ThermoFisher Scientific, 32132).
Liquid Chromatography-Mass spectrometry (LC-MS) analysis
Dried peptide samples were reconstituted in 20 µl LC-MS sample buffer (2% acetonitrile, 0.1% formic acid). Four µl of each sample were subjected to reverse phase liquid chromatography for peptide separation using an RSLCnano Ultimate 3000 system (ThermoFisher Scientific). Therefore, peptides were loaded on an Acclaim PepMap 100 pre-column (100 µm x 2 cm, C18, 5 µm, 100 Å; ThermoFisher Scientific, 164750) with 0.07% trifluoroacetic acid at a flow rate of 20 µL/min for 3 min. Analytical separation of peptides was done on an Acclaim PepMap RSLC column (75 µm x 50 cm, C18, 2 µm, 100 Å; ThermoFisher Scientific) at a flow rate of 300 nL/min. The solvent composition was gradually changed within 94 min from 96% solvent A (0.1% formic acid) and 4% solvent B (80% acetonitrile, 0.1% formic acid) to 10% solvent B within 2 minutes, to 30% solvent B within the next 58 min, to 45% solvent B within the following 22 min, and to 90% solvent B within the last 12 min of the gradient. All solvents and acids had Optima grade for LC-MS (ThermoFisher Scientific). Eluting peptides were on-line ionized by nano-electrospray (nESI) using the Nanospray Flex Ion Source (ThermoFisher Scientific) at 1.5 kV (liquid junction) and transferred into a Q Exactive HF mass spectrometer (ThermoFisher Scientific). Full scans in a mass range of 300 to 1650 m/z were recorded at a resolution of 60,000 followed by data-dependent top 10 HCD fragmentation at a resolution of 15,000 (dynamic exclusion enabled). LC-MS method programming and data acquisition was performed with the XCalibur 4.0 software (ThermoFisher Scientific).
Data analysis
Protein and biotin-site identification as well as SILAC-based quantification was performed with the MaxQuant 1.5.1.0 software (Max Planck Institute of Biochemistry) against an UniProt-derived S. cerevisiae specific database (http://www.uniprot.org, Proteome ID UP000002311, download 2019). The parameters were set according to Schmitt & Valerius (2019), with a maximum number of missed cleavage sites set to two [Citation58]. Generated data were then further processed and filtered with the Perseus software. Additionally, the Proteome DiscovererTM 2.2 software (ThermoFisher Scientific) was also used to identify candidates. The SequestHT and Mascot algorithms were used for database searches against a database specific for S. cerevisiae (SGD, 6110 entries, including common contaminants, S288C_ORF_database release version 2011, Stanford University).
Coimmunoprecipitation
The µMACS (magnetic activated cell sorting) System from Miltenyi Biotec was used for Co-IPs according to the manufacturer’s protocol (µMACSTM GFP Isolation Kit, Miltenyi Biotec, 130–091-125). Buffers were adjusted for optimal results: lysis buffer – 50 mM Tris HCl, pH 7.5, 100 mM NaCl, 1 mM EDTA, 0.5% (w:v) Tween-20 (Sigma-Aldrich, 7949), 1 mM PMSF, 1x 25x c0mplete, 0.1% (w:v) aprotinin (Merck, 616370), 0.1% (w:v) pepstatin (Merck, 516481), 0.1% (w:v) leupeptin (Merck, 108975), 0.1% (w:v) chymostatin (Merck, 230790); wash buffer 1–50 mM Tris HCl, pH 7.5, 100 mM NaCl, 0.025% (w:v) SDS; wash buffer 2–50 mM Tris HCl, pH 7.5. Yeast cultures expressing GFP-tagged bait and HA-tagged prey were grown to OD600 2–3, 200 OD600 units were harvested for the Co-IP. Cells were washed once with wash buffer 2, resuspended in lysis buffer with 400 µl glass beads and harshly shaken for 30 min at 4°C. Cell debris was removed (10,000 x g, 10 min, 4°C) and the supernatant was incubated with 50 µl GFP magnetic beads (Miltenyi Biotec, 130–091-125) for 30 min on ice. Beads were then isolated and washed three times with lysis buffer, once with wash buffer 1 and once with wash buffer 2. Proteins were eluted with in 2x Lämmli buffer as described above. Co-IPs were then analyzed by SDS-PAGE and western blotting. GFP and HA were detected as described above.
Fluorescence microscopy
Fluorescence microscopy was performed with a DeltaVision microscope (Olympus IX71, Applied Precision, Issaquah, USA) equipped with the UPlanSApo x100,1.4 numerical aperture (NA), oil immersion objective, a CoolSNAPHQ2TM couple-charged device (CCD) camera and different filter sets specific for GFP and mCherry. Imaging occurred with a 100x objective and 2 × 2 binning. At least 20 focal planes along the z-axis with a distance of 0.2 μm were captured and the resulting images were deconvolved using softWoRxTM Version 5.5.0 release 9 (Applied Precision, Issaquah, USA) and further processed with Fiji [Citation62].
Split-ubiquitin assay
One OD600 of cells was diluted 1:10, 1:100, 1:1,000 and 1:10,000 in sterile ddH2O, 4 µl were dropped on selection plates, on plates with 50 µg/ml uracil (Sigma-Aldrich, U1128) and 1 mg/ml 5-FOA (Fermentas, R0812). With the exception of the first plate, all were supplemented with 250 µM methionine and 100 µM CuSO4. Plates were grown up to 5 days at 30°C [Citation5,Citation37].
Statistical analysis
Blots were quantified using the free software Fiji [Citation62]. Statistical analyses for Western blots as well as fluorescence microscopy were performed using GraphPad Prism (GraphPad Software, USA). Graphs were plotted using the mean value together with the standard error of the mean (SEM). Statistical relevance was determined using the unpaired two-tailed t-test and is indicated in the graphs as follows: not significant (n.s. or no asterisk) for p > 0.05, * for p < 0.05, ** for p < 0.01, *** for p < 0.001 and **** for p < 0.0001.
Hyperosmotic vacuole fragmentation and FM 4-64 staining
We followed the previously established protocol as detailed in [Citation21]. FMTM 4–64 was purchased from Invitrogen (T3166).
Supplemental Material
Download MS Word (3.4 MB)Acknowledgments
We are grateful to P. Schlotterhose for technical support.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/15548627.2022.2072656
Additional information
Funding
References
- Wen X, Yang Y, Klionsky DJ. Moments in autophagy and disease: past and present. Mol Aspects Med. 2021:100966.
- Gomez-Sanchez R, Tooze SA, Reggiori F. Membrane supply and remodeling during autophagosome biogenesis. Curr Opin Cell Biol. 2021;71:112–119.
- Hollenstein DM, Kraft C. Autophagosomes are formed at a distinct cellular structure. Curr Opin Cell Biol. 2020;65:50–57.
- Matoba K, Noda NN. Structural catalog of core Atg proteins opens new era of autophagy research. J Biochem. 2021;169:517–525.
- Juris L, Montino M, Rube P, et al. PI3P binding by Atg21 organises Atg8 lipidation. EMBO J. 2015;34:955–973.
- Munzel L, Neumann P, Otto FB, et al. Atg21 organizes Atg8 lipidation at the contact of the vacuole with the phagophore. Autophagy. 2020;14:1–21.
- Hollenstein DM, Gomez-Sanchez R, Ciftci A, et al. Vac8 spatially confines autophagosome formation at the vacuole in S. cerevisiae. J Cell Sci. 2019;132:jcs235002.
- Gatica D, Wen X, Cheong H, et al. Vac8 determines phagophore assembly site vacuolar localization during nitrogen starvation-induced autophagy. Autophagy. 2021;17(7):1636–1648.
- Sawa-Makarska J, Baumann V, Coudevylle N, et al. Reconstitution of autophagosome nucleation defines Atg9 vesicles as seeds for membrane formation. Science (New York, NY). 2020;369:eaaz7714–12.
- Harada K, Kotani T, Kirisako H, et al. Two distinct mechanisms target the autophagy-related E3 complex to the pre-autophagosomal structure. eLife. 2019;8:685.
- Meiling-Wesse K, Barth H, Voss C, et al. Atg21 is required for effective recruitment of Atg8 to the preautophagosomal structure during the Cvt pathway. J Biol Chem. 2004;279:37741–37750.
- Stromhaug P, Reggiori F, Guan J, et al. Atg21 is a phosphoinositide binding protein required for efficient lipidation and localization of Atg8 during uptake of aminopeptidase I by selective autophagy. Mol Biol Cell. 2004;15:3553–3566.
- Noda NN. Atg2 and Atg9: intermembrane and interleaflet lipid transporters driving autophagy. biochimica et biophysica acta molecular and cell biology of lipids. Biochimica Et Biophysica Acta. Molecular and Cell Biology of Lipids. 2021;1866:158956.
- Yamamoto H, Kakuta S, Watanabe TM, et al. Atg9 vesicles are an important membrane source during early steps of autophagosome formation. J Cell Biol. 2012;198:219–233.
- Lei Y, Tang D, Liao G, et al. The crystal structure of Atg18 reveals a new binding site for Atg2 in Saccharomyces cerevisiae. Cell Mol Life Sci. 2020;290: 1717–13.
- Scacioc A, Schmidt C, Hofmann T, et al. Structure based biophysical characterization of the PROPPIN Atg18 shows Atg18 oligomerization upon membrane binding. Sci Rep. 2017;7:14008.
- Krick R, Busse RA, Scacioc A, et al. Structural and functional characterization of the two phosphoinositide binding sites of PROPPINs, a β-propeller protein family. Proc Natl Acad Sci U S A. 2012;109:E2042–9.
- Baskaran S, Ragusa MJ, Boura E, et al. Two-site recognition of phosphatidylinositol 3-phosphate by PROPPINs in autophagy. Mol Cell. 2012;47:339–348.
- Watanabe Y, Kobayashi T, Yamamoto H, et al. Structure-based analyses reveal distinct binding sites for Atg2 and phosphoinositides in Atg18. J Biol Chem. 2012;287:31681–31690.
- Nishimura T, Tooze SA. Emerging roles of ATG proteins and membrane lipids in autophagosome formation. Cell Discov. 2020;6: 32–18.
- Krick R, Henke S, Tolstrup J, et al. Dissecting the localization and function of Atg18, Atg21 and Ygr223c. Autophagy. 2008;4:896–910.
- Dove S, Piper R, McEwen R, et al. Svp1p defines a family of phosphatidylinositol 3,5-bisphosphate effectors. EMBO J. 2004;23:1922–1933.
- Reggiori F, Tucker KA, Stromhaug PE, et al. The Atg1-Atg13 complex regulates Atg9 and Atg23 retrieval transport from the pre-autophagosomal structure. Dev Cell. 2004;6:79–90.
- Efe JA, Botelho RJ, Emr SD. Atg18 regulates organelle morphology and Fab1 kinase activity independent of its membrane recruitment by phosphatidylinositol 3,5-bisphosphate. Mol Biol Cell. 2007;18:4232–4244. DOI:10.1091/mbc.e07-04-0301
- Jin N, Chow CY, Liu L, et al. VAC14 nucleates a protein complex essential for the acute interconversion of PI3P and PI(3,5)P(2) in yeast and mouse. EMBO J. 2008;27:3221–3234.
- Zieger M, Mayer A, Gruenberg JE. Yeast vacuoles fragment in an asymmetrical two-phase process with distinct protein requirements. Mol Biol Cell. 2012;23:3438–3449.
- Gopaldass N, Fauvet B, Lashuel H, et al. Membrane scission driven by the PROPPIN Atg18. EMBO J. 2017;36:e201796859–18.
- De Leo MG, Berger P, Mayer A. WIPI1 promotes fission of endosomal transport carriers and formation of autophagosomes through distinct mechanisms. Autophagy. 2021;17(11):3644–3670.
- Roux KJ, Kim DI, Burke B. BioID: a Screen for Protein-Protein Interactions. Hoboken NJ USA: John Wiley & Sons, Inc; 2001.
- Opitz N, Schmitt K, Hofer-Pretz V, et al. Capturing the Asc1p/receptor for activated C kinase 1 (RACK1) microenvironment at the head region of the 40S ribosome with quantitative BioID in Yeast. Mol Cell Proteomics. 2017;16:2199–2218.
- Ma M, Burd CG. Retrograde trafficking and plasma membrane recycling pathways of the budding yeast Saccharomyces cerevisiae. Traffic. Vol. 21. Copenhagen, Denmark 2020.45–59
- Seaman MNJ. The retromer complex: from genesis to revelations. Trends Biochem Sci. 2021;46:608–620.
- Bean BDM, Davey M, Conibear E. Cargo selectivity of yeast sorting nexins. Traffic. 2017;18(2):110–122.
- Efe JA, Botelho RJ, Emr SD. The Fab1 phosphatidylinositol kinase pathway in the regulation of vacuole morphology. Curr Opin Cell Biol. 2005;17:402–408.
- Meiling-Wesse K, Barth H, Thumm M. Ccz1p/Aut11p/Cvt16p is essential for autophagy and the cvt pathway. FEBS Lett. 2002;526:71–76.
- Welter E, Thumm M, Krick R. Quantification of nonselective bulk autophagy in S. cerevisiae using Pgk1-GFP. Autophagy. 2010;6:794–797.
- Müller J, Johnsson N. Split-ubiquitin and the split-protein sensors: chessman for the endgame. ChemBioChem. 2008;9:2029–2038.
- Purushothaman LK, Arlt H, Kuhlee A, et al. Retromer-driven membrane tubulation separates endosomal recycling from Rab7/Ypt7-dependent fusion. Mol Biol Cell. 2017;28:783–791.
- Liu Tt L, Gomez TS, Sackey BK, et al. Rab GTPase regulation of retromer-mediated cargo export during endosome maturation. Mol Biol Cell. 2012;23:2505–2515.
- Balderhaar HJK, Arlt H, Ostrowicz C, et al. The Rab GTPase Ypt7 is linked to retromer-mediated receptor recycling and fusion at the yeast late endosome. J Cell Sci. 2010;123:4085–4094.
- Popelka H, Damasio A, Hinshaw JE, et al. Structure and function of yeast Atg20, a sorting nexin that facilitates autophagy induction. Proc Natl Acad Sci U S A. 2017;114:E10112–E21.
- Ma M, Kumar S, Purushothaman L, et al. Lipid trafficking by yeast Snx4 family SNX-BAR proteins promotes autophagy and vacuole membrane fusion. Mol Biol Cell. 2018;29:2190–2200.
- Suzuki SW, Emr SD. Membrane protein recycling from the vacuole/lysosome membrane. J Cell Biol. 2018;256:jcb.201709162–15.
- Segarra VA, Boettner DR, Lemmon SK. Atg27 tyrosine sorting motif is important for its trafficking and Atg9 localization. Traffic. 2015;16:365–378.
- Ohashi Y, Munro S, Glick BS. Membrane delivery to the yeast autophagosome from the Golgi-endosomal system. Mol Biol Cell. 2010;21:3998–4008.
- Shirahama-Noda K, Kira S, Yoshimori T, et al. TRAPPIII is responsible for vesicular transport from early endosomes to Golgi, facilitating Atg9 cycling in autophagy. J Cell Sci. 2013;126:4963–4973.
- Ravussin A, Brech A, Tooze SA, et al. The phosphatidylinositol 3-phosphate-binding protein SNX4 controls ATG9A recycling and autophagy. J Cell Sci. 2021;134(3):jcs250670.
- Zavodszky E, Seaman MNJ, Moreau K, et al. Mutation in VPS35 associated with Parkinson’s disease impairs WASH complex association and inhibits autophagy. Nat Commun. 2014;5: 3828–16.
- Tamura N, Oku M, Ito M, et al. Atg18 phosphoregulation controls organellar dynamics by modulating its phosphoinositide-binding activity. J Cell Biol. 2013;202(4):685–698.
- Thumm M, Egner R, Koch B, et al. Isolation of autophagocytosis mutants of Saccharomyces cerevisiae. FEBS Lett. 1994;349:275–280.
- Janke C, Magiera M, Rathfelder N, et al. A versatile toolbox for PCR-based tagging of yeast genes: new fluorescent proteins, more markers and promoter substitution cassettes. Yeast (Chichester. England). 2004;21:947–962.
- Gueldener U, Heinisch J, Koehler GJ, et al. A second set of loxP marker cassettes for Cre-mediated multiple gene knockouts in budding yeast. Nucleic Acids Res. 2002;30:e23.
- Güldener U, Heck S, Fielder T, et al. A new efficient gene disruption cassette for repeated use in budding yeast. Nucleic Acids Res. 1996;24:2519–2524.
- Longtine M, McKenzie A, Demarini D, et al. Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast (Chichester. England). 1998;14:953–961.
- Suzuki K, Kirisako T, Kamada Y, et al. The pre-autophagosomal structure organized by concerted functions of APG genes is essential for autophagosome formation. EMBO J. 2001;20:5971–5981.
- Lang T, Reiche S, Straub M, et al. Autophagy and the cvt pathway both depend on AUT9. J Bacteriol. 2000;182:2125–2133.
- Barth H, Thumm M. A genomic screen identifies AUT8 as a novel gene essential for autophagy in the yeast Saccharomyces cerevisiae. Gene. 2001;274:151–156.
- Schmitt K, Valerius O. yRACK1/Asc1 proxiOMICs-Towards Illuminating Ships Passing in the Night. Cells. 2019;8:1384.
- Wang X, Li X, Li Y. A modified coomassie brilliant blue staining method at nanogram sensitivity compatible with proteomic analysis. Biotechnol Lett. 2007;29:79–90.
- Pink M, Verma N, Rettenmeier AW, et al. CBB staining protocol with higher sensitivity and mass spectrometric compatibility. Electrophoresis. 2010;31:593–598.
- Rappsilber J, Mann M, Ishihama Y. Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nat Protoc. 2007;2:1896–1906.
- Schindelin J, Arganda-Carreras I, Frise E, et al. Fiji: an open-source platform for biological-image analysis. Nat Methods. 2012;9:676–682.