
ABSTRACT
VMP1 is an ER membrane protein with phospholipid scramblase activity that has a critical role in regulating phagophore expansion and autophagosome closure. VMP1 also regulates lipid droplet formation and lipoprotein secretion in cultured cells and zebrafish. In a recent study, we showed that mice with hepatic deletion of Vmp1 have impaired very-low-density lipoprotein (VLDL) secretion and develop nonalcoholic steatohepatitis (NASH) even when fed with regular chow diet. Mechanistically, deletion of Vmp1 leads to decreased hepatic phosphatidylcholine (PC) and phosphatidylethanolamine (PE) levels as well as altered PC and PE acyl chain compositions resulting in the accumulation of neutral lipid structures in the ER phospholipid bilayer and decreased pre-VLDL assembly. These studies provide novel mechanistic insights into the non-autophagic functions of VMP1 in regulating lipoprotein secretion.
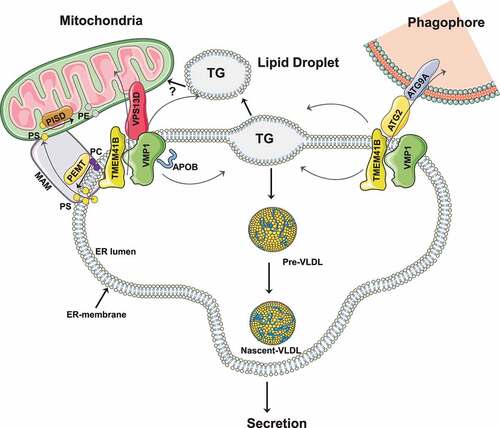
VMP1 (vacuole membrane protein 1) and its paralog protein TMEM41B (transmembrane protein 41B), which were recently identified as phospholipid scramblases critical for autophagy, are ER-resident multi-spanning membrane proteins that have a common downstream Escherichia coli DNA gene A (of hisT) (DedA) domain. While the exact mechanisms behind their roles in autophagy remain elusive, it has been proposed that VMP1 and TMEM41B may act upstream of lipid transfer proteins ATG2 and VPS13D as well as another scramblase, ATG9A, to form a lipid transport conduit to equilibrate phospholipid concentrations between the outer and inner leaflets of both ER and phagophore membranes, which is critical for phagophore expansion and autophagosome closure. Despite their similarity, deletion of the gene encoding either VMP1 or TMEM41B in cells is sufficient to block autophagic flux, and overexpression of VMP1 is able to correct the autophagy defect in TMEM41B-deficient cells but not vice versa, suggesting that VMP1 and TMEM41B may have non-redundant roles with VMP1 either predominant or downstream of TMEM41B in regulating autophagy.
Intriguingly, in addition to regulating autophagy, both VMP1 and TMEM41B have recently been shown to have non-autophagic functions in regulating lipoprotein secretion and NASH, a highly prevalent form of chronic liver disease. The liver is a major site for lipid metabolism, a process strictly regulated through a series of steps including fatty acid uptake, de novo lipogenesis, triglyceride esterification and storage, as well as fatty acid β-oxidation (FAO) and VLDL secretion. An imbalance between lipid input (intake and biosynthesis of triglyceride [TG]) and output (export and catabolism of TG) can result in NASH pathogenesis.
Assembly and secretion of VLDL, which is composed of a core of neutral lipids (mostly TG) surrounded by phospholipids and APOB/APOB100 (apolipoprotein B), starts in the endoplasmic reticulum (ER). Neutral lipids including TG, cholesterol ester and phospholipids are synthesized within the lipid bilayer of the ER, which are recruited by APOB into the ER lumen to form primordial VLDL (pre-VLDL). The pre-VLDL further recruits more neutral lipids to form nascent VLDL that is shuttled from the ER to the Golgi for further modification, and ultimately secretion from hepatocytes. In a recent study, we showed that deletion of Vmp1 in mouse livers severely impairs VLDL secretion resulting in rapid development of NASH features in mice, including steatosis, hepatocyte death, inflammation and fibrosis [Citation1]. More importantly, we found that levels of hepatic VMP1 decrease in liver samples of both humans with NASH and mice with diet-induced NASH, highlighting the relevance of our findings in human pathophysiology.
How does VMP1 regulate hepatic VLDL secretion? Three critical processes in VLDL secretion occur at the ER, including import of neutral lipids from the ER bilayer into the ER lumen, assembly of pre-VLDL in the ER lumen, and export of pre-VLDL from the ER lumen. In vmp1 KO hepatocytes, results from confocal microscopy studies showed that lipid droplets (LDs) are APOB and GFP-KDEL (an ER marker) positive but PLIN2 (perilipin 2; cytosolic LD marker) negative. Electron microscopy studies further revealed that most of these LDs are enclosed within the ER phospholipid bilayer and prevented from entering into the ER lumen.
Phospholipids, in particular phosphatidylcholine (PC) and phosphatidylethanolamine (PE) as well as their acyl chain compositions, are critical in ER membrane curvature and remodeling. Lipidomics analysis showed markedly decreased hepatic PC and PE content in vmp1 KO mouse livers, which may change ER membrane curvature such that neutral lipid structure entrance into the ER lumen and pe-VLDL assembly are impaired. Furthermore, we found that VMP1 is located at sites of ER-mitochondria contact termed mitochondria associated membranes (MAMs) (unpublished observation). The MAM is the critical site for the transport of phosphatidylserine (PS) from the ER to mitochondria and for the conversion of PS to PE by PISD/PSD1 (phosphatidylserine decarboxylase) in mitochondria. PEMT (phosphatidylethanolamine N-methyltransferase), a key enzyme for PC synthesis from PE, is also located at the MAM. Loss of VMP1 has been shown to increase ER contact with other organelles in HeLa cells, though we found decreased ER-mitochondrial contact in vmp1 KO hepatocytes (unpublished observations). While the reasons for these different observations remain unclear, it is likely that decreased numbers of MAM structures due to the absence of VMP1 may lead to decreased PISD and PEMT activities resulting in decreased PC and PE synthesis in hepatocytes.
In addition to impaired VLDL secretion, vmp1 KO hepatocytes have decreased mitochondrial FAO, which may also contribute to the NASH development. While the major cause remains elusive, several potential mechanisms may contribute to impaired FAO in vmp1 KO hepatocytes. First, it is conceivable that decreased FAO could be simply due to the lack of availability of fatty acids for mitochondrial FAO as most neutral lipids are trapped in the ER membrane. Second, while VMP1 is mainly an ER membrane protein, VMP1 also is located at the MAM, with lack of VMP1 possibly resulting in mitochondrial dysfunction by affecting the amount and composition of phospholipid in the mitochondrial membrane. Third, it has been shown that TMEM41B, ATG2 and ATG9A may form a lipid transport conduit for lipid transport from cytosolic LDs to mitochondria for FAO. Although direct interactions of VMP1 with ATG2 and ATG9A have not been reported, VMP1 interacts with TMEM41B and another lipid transport protein, VPS13D.Therefore it is likely that VMP1-TMEM41B, ATG2, VPS13D and ATG9A may form either one large or several distinctive lipid transport conduits that carry lipids to mitochondria for FAO. Deletion of either Tmem41b or Vmp1 is sufficient to impair mitochondrial FAO, suggesting that VMP1 and TMEM41B may act on the same lipid conduit and that the presence of one cannot compensate for the loss of the other, which is different from their aforementioned roles in regulating autophagy.
It is worth noting that cells lacking ATG7 or ATG5 do not have increased numbers of cellular LDs and that liver-specific atg5 KO mice have normal VLDL secretion and are resistant to fasting-induced hepatic steatosis, suggesting that autophagy is dispensable for the regulation of lipid metabolism by VMP1 or TMEM41B. It is likely that cellular location of phospholipid scramblases (i.e, ER vs plasma membrane) and their specific interactions with other autophagy related proteins (i.e., ATG2 and ATG9A) and/or apolipoproteins (i.e., APOB) are critical factors that account for the unique functions of VMP1 and TMEM41B compared with other phospholipid scramblases in the cell. Taken together, VMP1 and TMEM41B are emerging players in regulating autophagy and lipid metabolism (). Future work is needed to further dissect whether these protein’s roles in autophagy and lipid metabolism are regulated by mechanisms related to or distinct from their scramblase activity.
Figure 1. Autophagy and non-autophagic functions of VMP1 and TMEM41B. VMP1 and TMEM41B interact with ATG2 and ATG9A to regulate lipid transport from the ER to phagophore membranes. VMP1 regulates PC and PE synthesis at MAMs likely by affecting MAM structures and stability. VMP1 interacts with APOB to regulate lipoprotein biogenesis and secretion. VMP1 and TMEM41B also transfer lipids from LDs to mitochondria by interacting with ATG2 and VPS13D through yet unknown mechanisms.
Grant support: R37 AA020518, R01 DK102142, R01 AG072895 (WXD), P20GM144269 & R56 DK129234 (HMN).
Author’s contributions
WXD, XJ, AC & HMN conceived and wrote the manuscript.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
Reference
- Jiang X, Fulte S, Deng F, et al. Lack of VMP1 impairs hepatic lipoprotein secretion and promotes nonalcoholic steatohepatitis. J Hepatol. 2022. DOI:10.1016/j.jhep.2022.04.010