
ABSTRACT
The protein TRIM5 is under intensive investigation related to its roles in antiviral defense, yet its underlying mechanisms of action remain elusive. In our study, we performed an unbiased identification of TRIM5-interacting partners and found proteins participating in a wide variety of cellular functions. We utilized this proteomics data set to uncover a role for TRIM5 in mitophagy, a mitochondrial quality control system that is impaired in multiple human diseases. Mitochondrial damage triggers the recruitment of TRIM5 to ER-mitochondria contact sites where TRIM5 colocalizes with markers of autophagosome biogenesis. Cells lacking TRIM5 are unable to carry out PRKN-dependent and PRKN-independent mitophagy pathways. TRIM5 knockout cells show reduced mitochondrial function and uncontrolled immune activation in response to mitochondrial damage; phenotypes consistent with a requirement for TRIM5 in mitophagy. Mechanistically, we found that TRIM5 is required for the recruitment of the autophagy initiation machinery to damaged mitochondria, where TRIM5 acts as a scaffold promoting interactions between protein markers of mitochondrial damage and the autophagy initiation machinery.
The protein TRIM5 is under intensive investigation related to its roles in antiviral defense, yet its underlying mechanisms of action remain elusive. In our study, we performed an unbiased identification of TRIM5-interacting partners and found proteins participating in a wide variety of cellular functions. We utilized this proteomics data set to uncover a role for TRIM5 in mitophagy, a mitochondrial quality control system that is impaired in multiple human diseases. Mitochondrial damage triggers the recruitment of TRIM5 to ER-mitochondria contact sites where TRIM5 colocalizes with markers of autophagosome biogenesis. Cells lacking TRIM5 are unable to carry out PRKN-dependent and PRKN-independent mitophagy pathways. TRIM5 knockout cells show reduced mitochondrial function and uncontrolled immune activation in response to mitochondrial damage; phenotypes consistent with a requirement for TRIM5 in mitophagy. Mechanistically, we found that TRIM5 is required for the recruitment of the autophagy initiation machinery to damaged mitochondria, where TRIM5 acts as a scaffold promoting interactions between protein markers of mitochondrial damage and the autophagy initiation machinery.
Our first indication that TRIM5 may play a role in mitophagy came from an analysis of proteins that we identified as TRIM5 interactors in a proximity biotinylation-based mass spectrometry screen [Citation1]. As expected, based on TRIM5's known activities in antiviral defense, we found that TRIM5 interacts with multiple proteins involved in innate immune regulation. However, we unexpectedly see a dramatic enrichment of mitochondrial proteins in the TRIM5 interactome. Among these mitochondrial TRIM5 interactors, we identified several proteins (e.g., NIPSNAP1, NIPSNAP2, and PHB2) that are reported to function as mitophagy “eat-me tags” by marking damaged mitochondria and facilitating their autophagic removal. Given TRIM5's previously known interactions with a subset of autophagy proteins, we reasoned that TRIM5 might contribute to mitophagy.
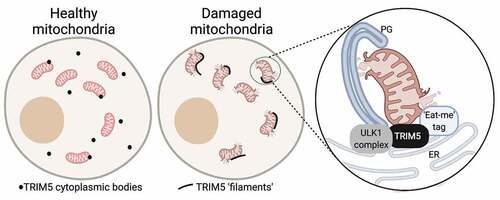
To test this idea, we first used imaging and mitochondrial fractionation experiments to confirm that TRIM5 can localize to the outer mitochondrial membrane. Interestingly, we observed a dramatic shift in the subcellular morphology of TRIM5 structures in response to mitochondrial damage. Under basal conditions, TRIM5 localizes to spherical cytoplasmic structures that sometimes are in contact with mitochondria. Following mitochondrial damage, TRIM5 localizes to elongated filamentous structures that intimately associate with PRKN-decorated mitochondria (). These TRIM5 filaments colocalize with CANX, a marker of the endoplasmic reticulum (ER). Thus, TRIM5 can localize to sites of ER-mitochondria contact. Because these sites are considered to be the physical platform from whence mitophagosome biogenesis initiates, our findings suggest that TRIM5 may play an early role in mitophagy. Accordingly, we found that mitochondrial depolarization increases interactions between TRIM5 and NIPSNAP1, NIPSNAP2, or PHB2 in coimmunoprecipitation experiments. Together, these findings revealed a previously unappreciated association between TRIM5 and mitochondria and demonstrated that mitochondrial damage enhances TRIM5 interactions with mitochondria-localized proteins involved in mitophagy.
Figure 1. Following mitochondrial damage, TRIM5 localizes to ER-mitochondrial contact sites where it recruits the autophagy initiation machinery, including ULK1 complex proteins, to proteins tagging damaged mitochondria (eat-me tags). In the absence of TRIM5, the recruitment of autophagy proteins to damaged mitochondria is abrogated, mitophagy is impaired, and cells show elevated inflammatory responses to mitochondrial damage. Abbreviations: ER, endoplasmic reticulum; PG, phagophore. Figure was created using BioRender.
Mammalian mitophagy pathways are classified based on their requirement for the E3 ubiquitin ligase PRKN. We find TRIM5 to be an important contributor to both PRKN-dependent mitophagy and PRKN-independent mitophagy. For these experiments, we used the mitochondrial uncouplers CCCP or oligomycin+antimycin A (O/A) to induce PRKN-dependent mitophagy and the antihelminthic agent ivermectin to induce PRKN-independent mitophagy. Knockout of TRIM5 strongly attenuates the degradation of mitochondrial proteins MT-CO2 and VDAC1 in response to these mitochondrial damaging agents in multiple cell lines. Expression of WT TRIM5, but not a TRIM5 mutant lacking its ubiquitin ligase activity, is able to restore mitophagy in TRIM5 knockout cells. TRIM5 is also required for the clearance of mitochondrial nucleoids in cells following CCCP or O/A treatment and for the colocalization between mitochondria and the autophagosome marker LC3B. Interestingly, we noted that TRIM5 knockout displays a more modest impact on mitophagy in PRKN-overexpressing HeLa cells, indicating that the relative importance of TRIM5-dependent mitophagy might be higher in more physiologically relevant model systems and suggesting that TRIM5 and PRKN may perform parallel roles in mitophagy. Mitophagy is important for maintaining mitochondrial function and for preventing inflammatory cellular responses to mitochondrial components (e.g., mitochondrial DNA). In agreement with its role in mitophagy, we showed that TRIM5 knockout decreases overall mitochondrial health while enhancing cell death responses and inflammatory gene expression (e.g., pro-IL1B/IL1β, OAS2, and IFIT1) in response to ivermectin treatment. Taken together, our data show a role for TRIM5 in mitophagy. The precise function of TRIM5's ubiquitin ligase activity in TRIM5-dependent mitophagy remains an unanswered question.
How does TRIM5 carry out its mitophagy functions? Whereas both CCCP and ivermectin treatment strongly enrich the presence of the mammalian Atg1 complex components ULK1, ATG13, and RB1CC1 in mitochondrial fractions in wild-type cells, we observe little to no recruitment of any of these proteins to mitochondria in TRIM5 knockout cells. This finding demonstrates that TRIM5 is an early actor in mitophagy that facilitates the recruitment of ULK1 complexes to damaged mitochondria. Accordingly, we observed that mitochondrial damage increases interactions between TRIM5 and ATG13 or RB1CC1 in coimmunoprecipitation experiments. Given that TRIM5 can also interact with “eat-me” tag proteins (e.g., NIPSNAP2), we next checked if eat-me proteins are in complexes with ULK1 complex components. Indeed, we are able to coimmunoprecipitate ATG13 and NIPSNAP2 or PHB2, especially following mitochondrial damage. TRIM5 expression strongly enhances ATG13-NIPSNAP2 and ATG13-PHB2 interactions. Collectively, our data show that TRIM5 is an assembly platform required for the recruitment of upstream autophagy regulators to eat-me tag proteins displayed on damaged mitochondria.
While previous research has focused exclusively on TRIM5's actions in antiviral defense, our study establishes that TRIM5 also has important homeostatic functions in mitochondrial quality control. However, because innate immunity can perceive damaged or dysfunctional mitochondria to be intracellular pathogens, the existence of proteins like TRIM5 that can simultaneously orchestrate direct pathogen restriction, innate immune signaling, and mitophagy should not be surprising. Furthermore, our identification of the TRIM5 interactome may yet lead to the discovery of additional new roles for this protein.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
Reference
- Saha B, Salemi M, Williams GL, et al. Interactomic analysis reveals a homeostatic role for the HIV restriction factor TRIM5alpha in mitophagy. Cell Rep. 2022;39(6):110797.