
ABSTRACT
The COVID-19 pandemic has caused substantial losses worldwide in people’s lives, health, and property. Currently, COVID-19 is still prominent worldwide without any specific drug treatment. The SARS-CoV-2 pathogen is the cause of various systemic diseases, mainly acute pneumonia. Within the pathological process, neutrophils are recruited to infected sites, especially in the lungs, for the first stage of removing invading SARS-CoV-2 through a range of mechanisms. Macroautophagy/autophagy, a conserved autodegradation process in neutrophils, plays a crucial role in the neutrophil phagocytosis of pathogens. NETosis refers to neutrophil cell death, while auto-inflammatory factors and antigens release NETs. This review summarizes the latest research progress and provides an in-depth explanation of the underlying mechanisms of autophagy and NETosis in COVID-19. Furthermore, after exploring the relationship between autophagy and NETosis, we discuss potential targets and treatment options. This review keeps up with the latest research on COVID-19 from neutrophil autophagy and NETosis with a new perspective, which can guide the urgent development of antiviral drugs and provide guidance for the clinical treatment of COVID-19.
Abbreviations: AKT1: AKT serine/threonine kinase 1; AMPK: AMP-activated protein kinase; AP: autophagosome; ARDS: acute respiratory distress syndrome; ATG: autophagy related; BECN1: beclin 1; cfDNA: cell-free DNA; COVID-19: coronavirus disease 2019; CQ: chloroquine; DMVs: double-membrane vesicles; ELANE/NE: elastase, neutrophil expressed; F3: coagulation factor III, tissue factor; HCQ: hydroxychloroquine; MAP1LC3/LC3: microtubule associated protein 1 light chain of 3; MPO: myeloperoxidase; MTORC1: mechanistic target of rapamycin kinase complex 1; NETs: neutrophil traps; NSP: nonstructural protein; PI3K: class I phosphoinositide 3-kinase; PtdIns3K: class III phosphatidylinositol 3-kinase; PtdIns3P: phosphatidylinositol-3-phosphate; ROS: reactive oxygen species; SARS-CoV-2: severe acute respiratory syndrome coronavirus 2; SKP2: S-phase kinase associated protein 2; TCC: terminal complement complex; ULK1: unc-51 like
Introduction
The widespread coronavirus 2 of the severe acute respiratory syndrome (SARS-CoV-2) has led to the coronavirus disease 2019 (COVID-19) epidemic, which has had a significant health and economic impact worldwide. Globally, 510 million people have been diagnosed with COVID-19, including 6 million deaths before May 2022 [Citation1]. Initial patients with COVID-19 in Wuhan described symptoms that included fever, cough, difficulty breathing, headache, tachycardia, progressive hypoxemia, and other signs and symptoms [Citation2]. In addition, acute respiratory distress syndrome (ARDS), heart damage, kidney damage, symptoms of multiple organ failure, and central nervous system damage are severe complications of COVID-19 that can lead to death [Citation3–6]. Currently, no effective targeted drugs have been developed, only symptomatic treatment for patients with COVID-19. Although vaccines have been developed, it is still urgent to find effective therapeutic targets and drugs. Exploring the mechanism of SARS-CoV-2 invading cells can help researchers find potentially effective therapeutic targets.
Neutrophils are essential for eliminating pathogens and have specific mechanisms against viruses [Citation7]. Neutrophils are the most abundant and effective defense against external pathogens, constituting the first line of cell defense [Citation8]. Neutrophils are recruited into the lungs in the first stage of COVID-19 and eliminate invading SARS-CoV-2 through numerous pathways. However, abnormal neutrophil activation causes a severe cytokine storm and host response in COVID-19 patients [Citation9]. Several clinical studies have shown that neutrophil levels in the blood are associated with a worsening of oxygenation in COVID-19 [Citation10,Citation11]. Neutrophils are the possible persistent inflammation and respiratory system damage factors in COVID-19 [Citation11].
The circulating platelet-neutrophil complexes in all patients with COVID-19 are elevated compared to the healthy individuals [Citation12]. COVID-19 infection induces immunothrombosis, in which activates neutrophil cells to interact with platelets and induce a coagulation cascade, resulting in the formation of intravascular clots in blood vessels [Citation13]. Neutrophils interact with endothelial cells and usually occur in neutrophil recruitment [Citation14]. Through endothelial cells, neutrophils translocate into the pulmonary microvasculature with activated platelets leading to several injuries such as epithelial layer damage, alveolar fibrin deposition, and further resulting in microthrombi formation [Citation15]. Emerging evidence indicates that endothelial dysfunction is involved in COVID-19 by changing the integrity of the vessel barrier, mediating neutrophilic infiltration and inflammation [Citation16], and contributing to end-organ damage [Citation17]. Therefore, these observations powerfully demonstrate that neutrophils could be considered one of the main therapeutic targets in COVID-19.
Macroautophagy/autophagy is a conserved autodegradation process in neutrophils activated under conditions that include pathogen infection, cell starvation, and endoplasmic reticulum stress [Citation18]. Autophagy is the only pathway that can produce random or targeted degradation of entire organelles and invade pathogens [Citation19]. First described by Mitroulis et al., autophagy is involved in phagocytosis-dependent and phagocytosis-independent initiation manners in human neutrophils [Citation20]. Autophagy is also essential for neutrophil differentiation [Citation21]. NETosis is a form of cell death caused primarily by neutrophils and refers to the formation of extracellular neutrophil traps (NETs) during this process [Citation22]. NETs consist of DNA fibers or nuclear chromatins that envelope cytosolic granule proteins and histones in a depolymerized reticulated form to trap and eliminate pathogens [Citation7,Citation23]. These two crucial cell death pathways play a significant role in the COVID-19 process; exploring these two neutrophil pathways may help researchers find suitable targets for treating COVID-19.
In this review, we discuss neutrophil autophagy and NETosis, their connection, and their contribution to the COVID-19 pathogenesis. Furthermore, this review also focuses on potential therapeutic targets and biomarkers of COVID-19 on autophagy and NETosis of neutrophils to guide the clinical development of anti-SARS-CoV-2 virus drugs to improve the prognosis and provide effective indicators to judge the severity of COVID-19.
Neutrophil autophagy in COVID-19
Autophagy of neutrophils in COVID-19: mechanisms
Autophagy plays a vital role in neutrophil formation, differentiation, and function. In addition, it plays a critical role in presenting pathogenic antigens, the secretion of related cytokines, degranulation, bactericidal activity, and inflammation control [Citation24,Citation25]. During pathogen invasion, neutrophils are activated and migrate to the lesion to phagocytose pathogens and produce effector molecules that kill pathogenic microorganisms, such as reactive oxygen species (ROS), proteinases, and NETs. All of these effector molecules can activate neutrophil autophagy [Citation8]. Through autophagy, harmful, redundant, and damaged cellular components are degraded. This process also provides neutrophils with essential lipids, amino acids, carbohydrates, and sufficient energy. Therefore, cell adaptability is improved under pathogen invasion and various pathophysiological conditions [Citation19].
During SARS-CoV-2 invasion, autophagy is activated and plays a vital role in the pathological process of COVID-19. The neutrophil autophagy mechanism for COVID-19 is described in . Neutrophil cell structures, such as damaged organelles caused by SARS-CoV-2 and cytoplasmic protein aggregates, are engulfed in phagophores that mature into double-membrane vesicles (DMVs) termed autophagosomes (APs) that are signs of autophagy under the control of proteins encoded by ATG (autophagy related) [Citation26]. These ATGs regulate autophagy in the following four processes: In the initial stage, neutrophils release ROS, proteases, and NET to eliminate the invading SARS-CoV-2, which can cause stress on cellular energy and increase the level of cytoplasmic AMP. Then, increasing AMP leads to the activation of AMP-activated protein kinase (AMPK), phosphorylation, and inactivation of the MTOR (mechanistic target of rapamycin kinase) complex 1 (MTORC1) [Citation27]. The next stage is nucleation controlled by the ULK1/Atg1 (unc-51 like autophagy activating kinase 1) complex downstream of MTORC1 [Citation28]. The ULK1/Atg1 complex activates the class III phosphatidylinositol 3-kinase (PtdIns3K) complex and produces an area rich in phosphatidylinositol-3-phosphate (PtdIns3P) on the surface of omegasomes. WIPI (WD repeat domain, phosphoinositide interacting) proteins recruit the ATG16L1 complex to promote the conversion of MAP1LC3/LC3 (microtubule associated protein 1 light chain 3)-I to the phosphatidylethanolamine-conjugated form, LC3-II [Citation29]. Then, in the expansion stage, autophagy is advanced by the two ubiquitin-like conjugation systems (ATG12–ATG5 and LC3/GABARAP/Atg8), as well as the PtdIns3K complex including ATG14-BECN1 (beclin 1)-PIK3C3/VPS34-PIK3R4/VPS15-NRBF2 [Citation29,]. Subsequently, BECN1 is degraded by SKP2 (S-phase kinase associated protein 2) and AKT1 (AKT serine/threonine kinase 1), a primary degradation pathway. The final stage is the maturation/degradation stage, in which APs and lysosomes fuse to form autolysosomes and the cargo sequestered within APs is degraded.
Figure 1. Autophagy and its mechanism in COVID-19. The autophagy process can be divided into four stages: (1) initiation, (2) nucleation, (3) expansion, and (4) maturation/degradation. In the initial stage, bioenergy stress in the host cell increases after SARS-CoV-2 invasion, thus increasing the level of cytoplasmic AMP: ATP, leading to activation of AMPK and phosphorylation/deactivation of MTORC1. Then in the nucleation stage, the ULK1 complex is activated, thus activating the PtdIns3K complex and producing a PtdIns3P-rich area (PtdIns3P) on the surface of omegasomes. PtdIns3P then recruits the WIPI and ZFYVE1 proteins that bind to the omegasome. In the expansion stage, ATG7 and ATG3 synergistically prime MAP1LC3/LC3, WIPI proteins recruit conjugation factors, including the ATG16L1 complex, leading to the conjugation of MAP1LC3/LC3 to phosphatidylethanolamine (generating LC3-II) to form APs. The last stage is maturation/decomposition, APs and lysosomes fuse to form autolysosomes. Substances wrapped in APs are degraded.
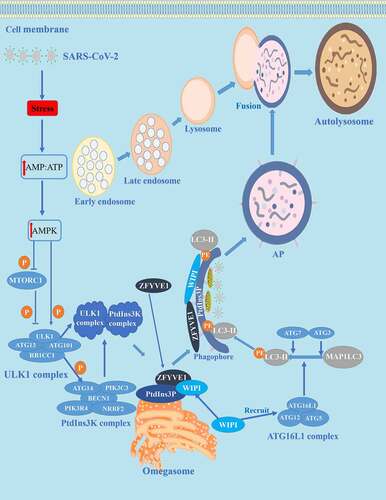
Coronavirus and neutrophil autophagy: a double-edged sword
Previous studies have shown that autophagy could be a double-edged sword during coronavirus infection [Citation30]. Autophagy can directly degrade coronavirus particles, promote the response to inflammatory cytokines [Citation31], and act as a negative inflammation regulator in cells such as neutrophils [Citation25]. Furthermore, autophagy promotes the presentation of antigens to induce immunity to resist coronavirus invasion [Citation32]. However, DMVs corresponding to APs (the morphological sign of autophagy) are beneficial in isolating the virus from the external immune response and thus become a site of virus replication and transcription [Citation31]. Previous studies have shown that several pathogens (e.g., influenza A virus, vesicular stomatitis virus, hepatitis B virus, hepatitis C virus, flaviviruses, coronaviruses) replicate in APs [Citation30,Citation33–35] and are capable of arresting autophagy by avoiding detection by the immune system. Nonstructural proteins (NSPs) are coronavirus replicase proteins. Among them, NSP3, NSP4 and NSP6 of SARS-CoV [Citation36], NSP2 and NSP3 of the equine arteritis virus [Citation37], and NSP3 and NSP4 of the middle eastern respiratory syndrome coronavirus [Citation38] induce rearrangement of the endoplasmic reticulum membrane to form DMVs and restrict AP expansion, producing small APs [Citation35].
Furthermore, SARS-CoV NSP6 colocalizes with LC3-positive DMVs [Citation39], indicating that autophagy is closely related to virus invasion. Coronaviruses also inhibit BECN1, which prevents the fusion of APs and lysosomes and interferon production [Citation40]. From this mechanism, coronaviruses control autophagy to facilitate its survival and replication in host cells.
The latest studies showed that SARS-CoV-2 inhibits complete autophagy and hinders AP maturation of APs to block autophagic flux in various ways. Mohamud and colleagues (2021) reported that the overexpression of papain-like protease of SARS-CoV-2 cleaves ULK1 and disrupts the formation of the ULK1-ATG13 complex to block complete host autophagy [Citation41]. Gassen and colleagues (2021) found that SARS-CoV-2 can restrict autophagy signal transduction by activating autophagy inhibitors (SKP2 and AKT1) and preventing AP-lysosome fusion [Citation42]. Network analysis indicated that SARS-CoV-2 could block autophagy flow by negatively regulating autophagy genes, SNAP29 (synaptosome associated protein 29), and lysosomal acidification genes in infected human nasopharyngeal samples [Citation43]. Therefore, the degradation of virus particles is reduced [Citation43]. Miao and colleagues (2021) showed that ORF3a of SARS-CoV-2 can isolate homotypic fusion and protein classification components, thus inhibiting AP and lysosome fusion of APs and lysosomes [Citation44]. ORF3a of SARS-CoV cannot interact with homotypic fusion and protein sorting parts to block autophagy, indicating that this is a newly discovered strategy and a unique characteristic of SARS-CoV-2 to inhibit autophagy [Citation44,Citation45].
In summary, the above studies suggest that SARS-CoV-2 can inhibit complete autophagy. However, Bouhaddou and colleagues (2020) found that the AMPK-MAPK14/p38 pathway is activated and the AKT pathway is inhibited, which can induce the autophagy in COVID-19 [Citation46]. Although that finding seems to contradict the above findings, incomplete autophagy is caused by the virus in DMVs interfering with the fusion of the APs with lysosomes in this circumstance, thus facilitating virus replication [Citation46].
NETosis of neutrophils in COVID-19
NETosis of neutrophils in COVID-19: mechanisms
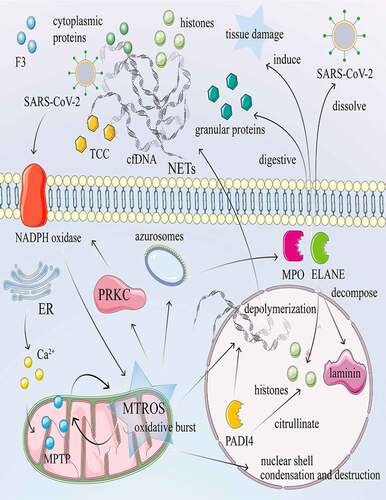
As a powerful defense method for the elimination and microbial destruction of pathogens, NETosis refers to neutrophil cell death of neutrophils, accompanied by the release of NETs, autoinflammatory factors, and antigens. NETs consist mainly of DNA fibers or nuclear chromatins that envelope cytosolic granule proteins and histones in a reticulated depolymerized form to trap and eliminate pathogens [Citation7,Citation23]. Neutrophils release ELANE (elastase, neutrophil expressed) and MPO (myeloperoxidase) from digestive granules, which can dissolve invading pathogens such as coronavirus, but the process can also cause tissue damage [Citation47]. Although the formation of NETs helps the host resist pathogens, excessive NET formation can trigger a series of inflammatory reactions, destroy surrounding tissues, and promote microthrombus formation [Citation9]. Excess NETs will stimulate the occurrence and development of many diseases and can damage many organs [Citation48].
The mechanism of neutrophil NETosis in COVID-19 is described in . There are two forms of NETosis, suicidal and vital [Citation49,Citation50]. Suicidal NETosis is a programmed cell death that ruptures the plasma membrane and releases NETs into the extracellular space. Under stressful conditions such as SARS-CoV-2 invasion, the NADPH oxidase complex is activated to generate large amounts of ROS [Citation51]. The azurosomes then dissociate under ROS, allowing ELANE and MPO to release the particles into the cytosol [Citation52]. Next, ELANE and MPO enter the nucleus to decompose laminin and histones, leading to condensation, destruction of the nuclear shell, and depolymerization of chromatin. PADI4 (peptidyl arginine deiminase 4) citrullinates histones and mediates chromatin decondensation. Decondensed deoxyribonucleic acid, released into the cytoplasm, binds to cytoplasmic proteins, extracellular histone H3, and granular proteins to create NET, which is eventually released extracellularly [Citation53]. In vital NETosis, NET is released without ROS production, and neutrophils retain their viability with normal function [Citation54]. Instead, lipopolysaccharides activate neutrophils and platelets through TLR4 (toll like receptor 4) [Citation50]. Furthermore, given that NETs are abundant in alveolar capillaries [Citation55], cell-free DNA (cfDNA) and NET histones can promote the release of pro-inflammatory cytokines and other comprehensive factors. Once NETs are released heavily in COVID-19, lung tissue is damaged by alveolar microcirculation disorders, and serious consequences can occur, such as ARDS [Citation56,Citation57].
Figure 2. NETosis and its mechanism in COVID-19. NETosis is a process of death with release of NET accompanied by auto-inflammatory factors and antigens. Calcium ionophore activates the CRAC channel of the cytoplasmic membrane and mobilizes Ca2+ in the endoplasmic reticulum, increasing the concentration of Ca2+ in the cytoplasm. Subsequently, Ca2+ overload and oxidative stress in the mitochondrial matrix lead to nonselective mitochondrial pores and the formation of MTROS. The release of MTROS into the cytoplasm activates PRKC and NADPH oxidase and induces the formation of NETs independently of MTROS and MPTP. Under stress conditions such as invasion of SARS-CoV-2, the NADPH oxidase complex is activated, subsequently producing large amounts of ROS. The azurosome is a protein complex that contains the ELANE and MPO proteins. Under ROS, the azurosome releases ELANE and MPO into the cytoplasm. In addition, the azurosomes dissociate in the presence of ROS, causing ELANE and MPO to enter the nucleus to decompose laminin and histones. PADI4 citrullinates histones, resulting in nucleocapsid agglomeration and chromatin depolymerization. DNA fibers or nuclear chromatin, released into the cytoplasm, combined with cytoplasmic proteins, granular proteins, histones, and F3, encapsulate the cytoplasm and finally in the extracellular form of NETs. cDNA and histone promote the release of pro-inflammatory cytokines and TCC. Neutrophils also release ELANE and MPO, which dissolve invasive pathogens such as SARS-CoV-2, digest particles, and induce tissue and bystander damage.
NETosis in COVID-19: clinical studies
SARS-CoV-2 has been shown to activate human neutrophil NETosis and is associated with increased intracellular ROS in neutrophils [Citation58]. There is evidence that low-density neutrophils, particularly prone to NETosis, are closely associated with blood vessel blockage and COVID-19-related ARDS [Citation57]. NET production increases in patients with acute respiratory failure and ARDS [Citation59]. Neutrophils and NETs were present in the airways and lung parenchyma alveoli in 40% of COVID-19 patients, indicating that the increase in NETosis is related to increased leukocytosis and neutrophils [Citation60]. Neutrophils increase in patients with COVID-19, suggesting the possibility of cytokine storms, while the reactivity of dendritic cells and monocytes is altered [Citation61].
NETosis is involved in developing many disease phenotypes (NET-mediated diseases) in COVID-19, including thrombosis, autoimmune diseases, sepsis, and multiorgan diseases such as lung diseases [Citation50,Citation62]. In thrombotic disorders, NETs are lined with F3 (coagulation factor III, tissue factor), the main initiator of blood coagulation [Citation63]. Clinical studies found that NET levels, F3 activity, and the terminal complement complex increased in serum from COVID-19 patients [Citation64]. Neutrophils from COVID-19 patients expressed high F3 and released NET with active F3 [Citation64]. The increased concentration of NETosis markers (cell-free DNA, MPO-DNA complexes, and histone H3) is correlated with neutrophil content and D-dimer (thrombosis marker) [Citation65,Citation66]. The NET formation can be stimulated by activated complement proteins that can interact with coagulation systems. NETs, in turn, can serve as a platform for complement activation and a scaffold for thrombus formation during coagulation [Citation67,Citation68]. Demonstration of autophagy-dependent extracellular delivery of F3 in NET further illustrates the role of neutrophils in thrombosis [Citation68].
Furthermore, components of NET, such as granule proteins and histones, can stimulate the production of antibodies and the development of autoimmune diseases [Citation69]. NET histones are the main contributors to cytotoxic effects toward endothelium injury and multiple organ dysfunction, further supporting the relevance of NET formation in sepsis [Citation62]. Inflammatory cytokines (such as IL1B/IL-1β) released by excessive NETosis are involved in ulcerative colitis and the development of cytokine storms, which are also the leading indicators of the severe course of COVID-19 [Citation9].
Autophagy and NETosis: relationship and potential targets in COVID-19
The relationship between autophagy and NETosis
Studies have shown that autophagy positively affects NET formation [Citation70]. Activation of autophagy (e.g., inhibition of MTOR through rapamycin) can enhance NET production [Citation64], while its inhibition (e.g., the use of a PtdIns3K inhibitor) can reduce NET release [Citation71], and decreased autophagy can decrease NET production [Citation72]. Furthermore, the class I phosphoinositide 3-kinase (PI3K)-AKT-MTOR pathway plays a significant role in both mechanisms, connecting autophagy and NET [Citation70]. Data from phorbol myristate acetate stimulation suggest that autophagy induction is an essential step for the initial stages of NETosis, but not for the cell death process itself [Citation73]. The formation of NETosis requires autophagy, which was investigated by removing WDFY3 to regulate ROS by NET reduction [Citation74]. Lack of either may result in the inability to form NETs and lead to cell apoptosis [Citation71]. In addition, studies have shown that ROS mediates the autophagy process [Citation75]. ROS can cause a rapid increase in the pH value of primary vesicles and induce autophagy [Citation76]. Although ROS is not essential for autophagy, ROS is indispensable for NETosis [Citation73]. Autophagy and superoxide generation is necessary but insufficient to mediate NETosis [Citation73].
Furthermore, several neutrophil-derived proteins, such as F3 and IL1B, are enclosed in the AP and, through these structures, are exposed to NETs [Citation68,Citation77]. Demonstration of autophagy-dependent extracellular delivery of F3 in NET further illustrates the role of neutrophils in thrombosis [Citation68]. Autophagy IL1B maturation and release of IL1B-bearing NETs are closely related to autoinflammatory disorders such as ulcerative colitis and Behçet’s disease [Citation77]. These observations suggest a strong link between autophagy NETs and disease phenotypes. Understanding the relationship between neutrophil autophagy and NETosis is critical for identifying potential targets in these two mechanisms.
Potential therapeutic targets for autophagy and NETosis in COVID-19
Although there is currently no specific drug treatment for COVID-19, regulation of autophagy and NETosis holds promise for developing drugs related to COVID-19. Potential therapeutic targets for autophagy and NETosis in COVID-19 are described in and .
Table 1. Potential therapeutic targets of autophagy in COVID-19.
Table 2. Potential therapeutic targets for NETosis in COVID-19.
Autophagy features resistance to SARS-CoV and SARS-CoV-2, immune evasion, and replication promotion, which is beneficial for the existence of the coronavirus. However, this practical regulation window is so narrow that both autophagy inhibitors and autophagy inducers can have an antiviral effect on COVID-19 [Citation78]. Although autophagy induction can enhance virus clearance and limit SARS-CoV-2 proliferation [Citation42], the promotion of NETosis is not conducive to treatment and can hinder recovery. Autophagy inhibitors could act indirectly to combat the spread of SARS-CoV-2. Due to the crosstalk between autophagy and apoptosis, the accumulation of APs could activate the host apoptosis mechanism, causing the death of host cells and thus stopping virus replication [Citation29].
Furthermore, inhibition of autophagy can inhibit NET expression, thus reducing inflammation and ARDS and other complications such as sepsis, mediterranean fever, and ulcerative colitis [Citation79–82]. Therefore, we do not recommend autophagy inducers as potential therapeutic drugs since autophagy-mediated NETs induce inflammation and exacerbate tissue damage [Citation79]. In contrast, we recommend autophagy inhibitors, as they also inhibit NETosis.
Among autophagy inhibitors, 3-methyladenine and wortmannin can block autophagy and inhibit virus replication [Citation83]. Chloroquine (CQ) and hydroxychloroquine (HCQ) are lysosomal nutrients that can alkalize lysosomes, inhibit AP and lysosome fusion, block protease activity, and inhibit virus entry. With broad-spectrum antiviral and immunomodulatory properties, these autophagy inhibitors are promising drugs to combat COVID-19 [Citation78,Citation84]. However, its use is controversial given its high cytotoxicity and side effects. Nitazoxanide can inhibit late-stage autophagy and has been shown to inhibit SARS-CoV-2 infection in vitro. Azithromycin is another potential drug for COVID-19, as it can hinder lysosomal acidification, thus inhibiting AP clearance and degradation [Citation85]. Furthermore, azithromycin can extensively block autophagy combined with CQ or HCQ to enhance the curative effect on COVID-19 [Citation86]. Finally, cepharanthine is the other primary drug that can inhibit autophagy with antiviral activity and may be a promising therapeutic option for COVID-19 [Citation87].
Multiple studies have confirmed that the endocytic pathway in autophagy is the primary pathway through which SARS-CoV-2 enters host cells. Therefore, the endocytic pathway has been widely studied as a target for treating COVID-19 [Citation28]. Chlorpromazine, an inhibitor of clathrin-dependent endocytosis and an antischizophrenia drug, has been shown to significantly inhibit the entry of coronavirus into host cells [Citation88]. As an FDA-approved anticoronavirus drug, it can be used alongside other medications to treat COVID-19, but its effectiveness has not been clinically evaluated [Citation89]. Furthermore, NH4Cl [Citation90], CQ [Citation28], and bafilomycin A1 [Citation91] are lysosomal nutrients that can inhibit virus entry and prevent virus infection. Ouabain and bufalin [Citation92] are drugs that can hinder the clathrin-mediated endocytic pathway to avoid the access of coronaviruses.
For NETosis, the MPO-DNA complex is a well-defined marker of NETosis [Citation93]. cfDNA, extracellular histone H3, growth arrest-specific 6, and ELANE, which increased significantly in patients with COVID-19, can be used as biomarkers for NETosis [Citation56,Citation65,Citation94]. NET expressions are positively correlated with thrombosis. Thus, inhibition of NET decreases platelet-mediated NET-driven thrombogenicity, which is beneficial for the recovery of COVID-19 [Citation64]. Accurate hematological markers of systemic inflammation, systemic inflammation index, and high neutrophil to lymphocyte ratio were closely related to the severity and mortality of COVID-19 patients [Citation95,Citation96]. Furthermore, circulating nucleosomes and citrulline levels can be used clinically as quick and effective measures of the severity of COVID-19 [Citation97].
From the theory of NETosis, cfDNA may be a potential therapeutic target for treating SARS-CoV-2-induced sepsis, and exogenous administration of long-acting nanoparticle deoxyribonuclease-1 may effectively reduce neutrophil activity and cfDNA levels as a therapeutic intervention for COVID-19 [Citation94]. Furthermore, as with other pathogens, SARS-CoV-2 activates platelets to release extracellular vesicles and activates the C-type lectin receptor that enhances NET formation [Citation98]. Therefore, blocking C-type lectins could be a promising therapeutic strategy to reduce intravascular coagulopathy and SARS-CoV-2-induced NETosis [Citation99]. Furthermore, immunomodulatory treatment is a future direction of clinical treatment of COVID-19, such as the treatment with anakinra, eculizumab, heparin, hyperbaric oxygen, remdesivir, and sirolimus [Citation49].
Furthermore, DNase treatment significantly reduces citrullination of histone H3 levels and degrades NET [Citation100]. Clinical studies showed that DNase-I-coated melanin-like nanospheres can reduce neutrophil counts and mitigate sepsis-associated NETosis dysregulation, thus alleviating systemic inflammation and attenuating mortality [Citation53]. Recent data based on a clinical study in the process (ClinicalTrials.gov Identifier: NCT05279391) to treat COVID-19 suggested that the treatment consisted of inhaled DNase-I to dissolve thrombogenic NETs and agents against cytokine-mediated hyperinflammation, was associated with lower in-hospital mortality and intubation rate, shorter duration of hospitalization, and prolonged overall survival [Citation101]. In addition, the treatment also has an inhibitory effect on the Thrombin axis in fibroblasts [Citation101]. Taken together, inhaled DNase-I has emerged as a potential therapeutic option and has potential applications in the treatment of COVID-19.
Concluding remarks
Autophagy and NETosis are two primary defense mechanisms in neutrophils with different functions during the SARS-CoV-2 invasion process. This review provides an in-depth explanation of the underlying mechanisms of autophagy and NETosis in COVID-19 and summarizes the latest research progress. After exploring the relationship between autophagy and NETosis, we also discussed potential targets and treatment options for COVID-19. A comprehensive understanding of neutrophil responses of patients with COVID-19 is required to understand the heterogeneity of the severity of the disease, define the effectiveness of treatment, and predict the prognosis. In this respect, a single-cell multi-omics approach would contribute to achieving this goal.
Statements and Declarations
The authors declare that they have no conflict of interest.
Data sharing statement
Not applicable.
Acknowledgments
Not applicable.
Disclosure statement
The authors declare that they have no conflict of interest.
Correction Statement
This article has been corrected with minor changes. These changes do not impact the academic content of the article.
Additional information
Funding
References
- World Health Organization. Coronavirus (COVID-19) Dashboard data table. [Cited 2022 May 1]. https://covid19.who.int/.
- Wang D, Hu B, Hu C, et al. Clinical characteristics of 138 hospitalized patients with 2019 novel coronavirus–infected pneumonia in Wuhan, China. Jama. 2020;323(11):1061–1069.
- Carvalho T, Krammer F, Iwasaki A. The first 12 months of COVID-19: a timeline of immunological insights. Nat Rev Immunol. 2021;21(4):245–256.
- Mallah SI, Ghorab OK, Al-Salmi S, et al. COVID-19: breaking down a global health crisis. Ann Clin Microbiol Antimicrob. 2021;20(1):35.
- Aleksova A, Gagno G, Sinagra G, et al. Effects of SARS-CoV-2 on cardiovascular system: the dual role of angiotensin-converting enzyme 2 (ACE2) as the virus receptor and homeostasis regulator-review. Int J Mol Sci. 2021:22. DOI:10.3390/ijms23010022.
- Zhang R, Sun C, Chen X, et al. COVID-19-related brain injury: the potential role of ferroptosis. J Inflamm Res. 2022;15:2181–2198.
- Borges L, Pithon-Curi TC, Curi R, et al. COVID-19 and neutrophils: the relationship between hyperinflammation and neutrophil extracellular traps. Mediators Inflamm. 2020;2020:8829674.
- Chargui A, El May MV. Autophagy mediates neutrophil responses to bacterial infection. Apmis. 2014;122(11):1047–1058.
- Barnes BJ, Adrover JM, Baxter-Stoltzfus A, et al. Targeting potential drivers of COVID-19: neutrophil extracellular traps. J Exp Med. 2020;217(6). DOI:10.1084/jem.20200652.
- Zhang B, Zhou X, Zhu C, et al. Immune phenotyping based on the neutrophil-to-lymphocyte ratio and IgG level predicts disease severity and outcome for patients with COVID-19. Front Mol Biosci. 2020;7:157.
- Shi H, Zuo Y, Yalavarthi S, et al. Neutrophil calprotectin identifies severe pulmonary disease in COVID-19. J Leukoc Biol. 2021;109(1):67–72.
- Manne BK, Denorme F, Middleton EA, et al. Platelet gene expression and function in patients with COVID-19. Blood. 2020;136(11):1317–1329.
- Bonaventura A, Vecchié A, Dagna L, et al. Endothelial dysfunction and immunothrombosis as key pathogenic mechanisms in COVID-19. Nat Rev Immunol. 2021;21(5):319–329.
- Filippi M-D. Neutrophil transendothelial migration: updates and new perspectives. Blood. 2019;133(20):2149–2158.
- Morris G, Bortolasci CC, Puri BK, et al. Preventing the development of severe COVID-19 by modifying immunothrombosis. Life Sci. 2021;264:118617.
- Jin Y, Ji W, Yang H, et al. Endothelial activation and dysfunction in COVID-19: from basic mechanisms to potential therapeutic approaches. Signal Transduct Target Ther. 2020;5(1):293.
- Evans PC, Rainger GE, Mason JC, et al. Endothelial dysfunction in COVID-19: a position paper of the ESC working group for atherosclerosis and vascular biology, and the ESC council of basic cardiovascular science. Cardiovasc Res. 2020;116(14):2177–2184.
- Miller K, McGrath ME, Hu Z, et al. Coronavirus interactions with the cellular autophagy machinery. Autophagy. 2020;16(12):2131–2139.
- Mijaljica D, Klionsky DJ. Autophagy/virophagy: a “disposal strategy” to combat COVID-19. Autophagy. 2020;16(12):2271–2272.
- Mitroulis I, Kourtzelis I, Kambas K, et al. Regulation of the autophagic machinery in human neutrophils. Eur J Immunol. 2010;40(5):1461–1472.
- Sidaway P. Neutrophils: neutrophil differentiation is autophagy dependent. Nat Rev Immunol. 2017;17(11):662.
- Nirmala JG, Lopus M. Cell death mechanisms in eukaryotes. Cell Biol Toxicol. 2020;36(2):145–164.
- Gupta S, Sahni V. The intriguing commonality of NETosis between COVID-19 & Periodontal disease. Med Hypotheses. 2020;144:109968.
- Shibutani ST, Saitoh T, Nowag H, et al. Autophagy and autophagy-related proteins in the immune system. Nat Immunol. 2015;16(10):1014–1024.
- Shrestha S, Lee JM, Hong C-W. Autophagy in neutrophils. Korean J Physiol Pharmacol. 2020;24(1):1–10.
- Mizushima N. The ATG conjugation systems in autophagy. Curr Opin Cell Biol. 2020;63:1–10.
- Galluzzi L, Green DR. Autophagy-independent functions of the autophagy machinery. Cell. 2019;177(7):1682–1699.
- Yang N, Shen H-M. Targeting the endocytic pathway and autophagy process as a novel therapeutic strategy in COVID-19. Int J Biol Sci. 2020;16(10):1724–1731.
- Sargazi S, Sheervalilou R, Rokni M, et al. The role of autophagy in controlling SARS-CoV-2 infection: an overview on virophagy-mediated molecular drug targets. Cell Biol Int. 2021;45(8):1599–1612.
- Choi Y, Bowman JW, Jung JU. Autophagy during viral infection — a double-edged sword. Nat Rev Microbiol. 2018;16(6):341–354.
- García-Pérez BE, González-Rojas JA, Salazar MI, et al. Taming the Autophagy as a strategy for treating COVID-19. Cells. 2020;9(12):2679.
- Ahmad L, Mostowy S, Sancho-Shimizu V. Autophagy-virus interplay: from cell biology to human disease. Front Cell Dev Biol. 2018;6:155.
- Wang R, Zhu Y, and Zhao J, et al. Autophagy Promotes Replication of Influenza A Virus In Vitro. J Virol. 2019;93(4):e01984–18.
- Lin Y, Wu C, Wang X, et al. Glucosamine promotes hepatitis B virus replication through its dual effects in suppressing autophagic degradation and inhibiting MTORC1 signaling. Autophagy. 2020;16(3):548–561.
- Cottam EM, Whelband MC, Wileman T. Coronavirus NSP6 restricts autophagosome expansion. Autophagy. 2014;10(8):1426–1441.
- Angelini MM, Akhlaghpour M, Neuman BW, et al. Severe acute respiratory syndrome coronavirus nonstructural proteins 3, 4, and 6 induce double-membrane vesicles. mBio. 2013;4(4). DOI:10.1128/mBio.00524-13
- Snijder EJ, van Tol H, Roos N, et al. Non-structural proteins 2 and 3 interact to modify host cell membranes during the formation of the arterivirus replication complex. J Gen Virol. 2001;82(5):985–994.
- Oudshoorn D, Rijs K, Limpens R, et al. Expression and cleavage of middle east respiratory syndrome coronavirus nsp3-4 polyprotein induce the formation of double-membrane vesicles that mimic those associated with coronaviral RNA replication. mBio. 2017;8(6). DOI:10.1128/mBio.01658-17.
- Cottam EM, Maier HJ, Manifava M, et al. Coronavirus nsp6 proteins generate autophagosomes from the endoplasmic reticulum via an omegasome intermediate. Autophagy. 2011;7(11):1335–1347.
- Gassen NC, Niemeyer D, Muth D, et al. SKP2 attenuates autophagy through Beclin1-ubiquitination and its inhibition reduces MERS-Coronavirus infection. Nat Commun. 2019;10(1):5770.
- Mohamud Y, Xue YC, Liu H, et al. The papain-like protease of coronaviruses cleaves ULK1 to disrupt host autophagy. Biochem Biophys Res Commun. 2021;540:75–82.
- Gassen NC, Papies J, Bajaj T, et al. SARS-CoV-2-mediated dysregulation of metabolism and autophagy uncovers host-targeting antivirals. Nat Commun. 2021;12(1):3818.
- Singh K, Chen Y-C, Hassanzadeh S, et al. Network analysis and transcriptome profiling identify autophagic and mitochondrial dysfunctions in SARS-CoV-2 infection. Front Genet. 2021;12:599261.
- Miao G, Zhao H, Li Y, et al. ORF3a of the COVID-19 virus SARS-CoV-2 blocks HOPS complex-mediated assembly of the SNARE complex required for autolysosome formation. Dev Cell. 2021;56(4):427–442.e5.
- Zhao Z, Lu K, Mao B, et al. The interplay between emerging human coronavirus infections and autophagy. Emerg Microbes Infect. 2021;10(1):196–205.
- Bouhaddou M, Memon D, Meyer B, et al. The Global Phosphorylation Landscape of SARS-CoV-2 Infection. Cell. 2020;182(3):685–712.e19.
- Sawadogo SA, Dighero-Kemp B, Ouédraogo -D-D, et al. How NETosis could drive “Post-COVID-19 syndrome” among survivors. Immunol Lett. 2020;228:35–37.
- Papayannopoulos V. Neutrophil extracellular traps in immunity and disease. Nat Rev Immunol. 2018;18(2):134–147.
- Cicco S, Cicco G, Racanelli V, et al. Neutrophil extracellular traps (NETs) and damage-associated molecular patterns (DAMPs): two potential targets for COVID-19 treatment. Mediators Inflamm. 2020;2020:7527953.
- Vorobjeva NV, Chernyak BV. NETosis: molecular mechanisms, role in physiology and pathology. Biochemistry (Mosc). 2020;85(10):1178–1190.
- Delgado-Rizo V, Martínez-Guzmán MA, Iñiguez-Gutierrez L, et al. Neutrophil extracellular traps and its implications in inflammation: an overview. Front Immunol. 2017;8:81.
- Metzler KD, Goosmann C, Lubojemska A, et al. A myeloperoxidase-containing complex regulates neutrophil elastase release and actin dynamics during NETosis. Cell Rep. 2014;8(3):883–896.
- Park HH, Park W, Lee YY, et al. Bioinspired DNase-I-coated melanin-like nanospheres for modulation of infection-associated NETosis dysregulation. Adv Sci (Weinh). 2020;7(23):2001940.
- Yousefi S, Simon D, Stojkov D, et al. In vivo evidence for extracellular DNA trap formation. Cell Death Dis. 2020;11(4):300.
- Chauhan AJ, Wiffen LJ, Brown TP. COVID-19: a collision of complement, coagulation and inflammatory pathways. J Thromb Haemost. 2020;18(9):2110–2117.
- Porto BN, Stein RT. Neutrophil extracellular traps in pulmonary diseases: too much of a good thing? Front Immunol. 2016;7:311.
- Obermayer A, Jakob L-M, Haslbauer JD, et al. Neutrophil extracellular traps in fatal COVID-19-associated lung injury. Dis Markers. 2021;2021:5566826.
- Arcanjo A, Logullo J, Menezes CCB, et al. The emerging role of neutrophil extracellular traps in severe acute respiratory syndrome coronavirus 2 (COVID-19). Sci Rep. 2020;10(1):19630.
- Bendib I, de Chaisemartin L, Granger V, et al. Neutrophil extracellular traps are elevated in patients with pneumonia-related acute respiratory distress syndrome. Anesthesiology. 2019;130(4):581–591.
- Masso-Silva JA, Moshensky A, and Lam MTY, et al. Increased peripheral blood neutrophil activation phenotypes and NETosis in critically ill COVID-19 patients: a case series and review of the literature. Clin Infect Dis. 2021;74(3):479–489.
- Parackova Z, Zentsova I, Bloomfield M, et al. Disharmonic inflammatory signatures in COVID-19: augmented neutrophils’ but impaired monocytes’ and dendritic cells’ responsiveness. Cells. 2020;9(10):2206.
- Hamam HJ, Palaniyar P. Post-translational modifications in NETosis and NETs-mediated diseases. Biomolecules. 2019;9(8):369.
- Carminita E, Crescence L, Panicot-Dubois L, et al. Role of neutrophils and NETs in animal models of thrombosis. Int J Mol Sci. 2022;23(3):1411.
- Skendros P, Mitsios A, Chrysanthopoulou A, et al. Complement and tissue factor–enriched neutrophil extracellular traps are key drivers in COVID-19 immunothrombosis. J Clin Invest. 2020;130(11):6151–6157.
- Huckriede J, Anderberg SB, Morales A, et al. Evolution of NETosis markers and DAMPs have prognostic value in critically ill COVID-19 patients. Sci Rep. 2021;11(1):15701.
- Bautista-Becerril B, Campi-Caballero R, Sevilla-Fuentes S, et al. Immunothrombosis in COVID-19: implications of neutrophil extracellular traps. Biomolecules. 2021;11(5):694.
- de Bont CM, Boelens WC, Pruijn GJM. NETosis, complement, and coagulation: a triangular relationship. Cell Mol Immunol. 2019;16(1):19–27.
- Kambas K, Mitroulis I, Ritis K. The emerging role of neutrophils in thrombosis—the journey of TF through NETs. Front Immunol. 2012;3:385.
- Gupta S, Kaplan MJ. The role of neutrophils and NETosis in autoimmune and renal diseases. Nat Rev Nephrol. 2016;12(7):402–413.
- Yu Y, Sun B. Autophagy-mediated regulation of neutrophils and clinical applications. Burns Trauma. 2020;8:tkz001.
- Itakura A, McCarty OJT. Pivotal role for the mTOR pathway in the formation of neutrophil extracellular traps via regulation of autophagy. Am J Physiol Cell Physiol. 2013;305(3):C348–54.
- Sharma A, Simonson TJ, Jondle CN, et al. Mincle-mediated neutrophil extracellular trap formation by regulation of Autophagy. J Infect Dis. 2017;215(7):1040–1048.
- Remijsen Q, Vanden Berghe T, Wirawan E, et al. Neutrophil extracellular trap cell death requires both autophagy and superoxide generation. Cell Res. 2011;21(2):290–304.
- Suzuki E, Maverakis E, Sarin R, et al. T cell–independent mechanisms associated with neutrophil extracellular trap formation and selective Autophagy in IL-17A–mediated epidermal hyperplasia. J Immunol. 2016;197(11):4403–4412.
- Karna P, Zughaier S, Pannu V, et al. Induction of reactive oxygen species-mediated autophagy by a novel microtubule-modulating agent. J Biol Chem. 2010;285(24):18737–18748.
- Yun HR, Jo YH, Kim J, et al. Roles of Autophagy in oxidative stress. Int J Mol Sci. 2020;21(9):3289.
- Skendros P, Papagoras C, Mitroulis I, et al. Autoinflammation: lessons from the study of familial mediterranean fever. J Autoimmun. 2019;104:102305.
- Bello-Perez M, Sola I, Novoa B, et al. Canonical and Noncanonical Autophagy as Potential Targets for COVID-19. Cells. 2020;9(7):1619.
- Liang X, Liu L, Wang Y, et al. Autophagy-driven NETosis is a double-edged sword – review. Biomed Pharmacother. 2020;126:110065.
- Mohammed BM, Fisher BJ, Kraskauskas D, et al. Vitamin C: a novel regulator of neutrophil extracellular trap formation. Nutrients. 2013;5(8):3131–3150.
- Skendros P, Chrysanthopoulou A, Rousset F, et al. Regulated in development and DNA damage responses 1 (REDD1) links stress with IL-1β–mediated familial Mediterranean fever attack through autophagy-driven neutrophil extracellular traps. J Allergy Clin Immunol. 2017;140(5):1378–1387.e13.
- Angelidou I, Chrysanthopoulou A, Mitsios A, et al. REDD1/Autophagy pathway is associated with neutrophil-driven il-1β inflammatory response in active ulcerative colitis. J Immunol. 2018;200(12):3950–3961.
- Hui X, Zhang L, Cao L, et al. SARS-CoV-2 promote autophagy to suppress type I interferon response. Signal Transduct Target Ther. 2021;6(1):180.
- Wang M, Cao R, Zhang L, et al. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res. 2020;30(3):269–271.
- Venditto VJ, Haydar D, Abdel-Latif A, et al. Immunomodulatory effects of azithromycin revisited: potential applications to COVID-19. Front Immunol. 2021;12:574425.
- Gautret P, Lagier J-C, Parola P, et al. Hydroxychloroquine and azithromycin as a treatment of COVID-19: results of an open-label non-randomized clinical trial. Int J Antimicrob Agents. 2020;56(1):105949.
- Li S, Liu W, Chen Y, et al. Transcriptome analysis of cepharanthine against a SARS-CoV-2-related coronavirus. Brief Bioinform. 2021;22(2):1378–1386.
- Otręba M, Kośmider L, Rzepecka-Stojko A. Antiviral activity of chlorpromazine, fluphenazine, perphenazine, prochlorperazine, and thioridazine towards RNA-viruses. A review. Eur J Pharmacol. 2020;887:173553.
- Stip E. Psychiatry and COVID-19: the role of chlorpromazine. Can J Psychiatry. 2020;65(10):739–740.
- Hidvégi M, Nichelatti M. Bacillus calmette-guerin vaccination policy and consumption of ammonium chloride-enriched confectioneries may be factors reducing COVID-19 death rates in Europe. Isr Med Assoc J. 2020;22:501–504.
- Zhang J, Chen J, Shi D, et al. Porcine deltacoronavirus enters cells via two pathways: a protease-mediated one at the cell surface and another facilitated by cathepsins in the endosome. J Biol Chem. 2019;294(25):9830–9843.
- Burkard C, Verheije MH, Haagmans BL, et al. ATP1A1-mediated Src signaling inhibits coronavirus entry into host cells. J Virol. 2015;89(8):4434–4448.
- Stakos D, Skendros P, Konstantinides S, et al. Traps N‘ clots: NET-mediated thrombosis and related diseases. Thromb Haemost. 2020;120(3):373–383.
- Lee YY, Park HH, Park W, et al. Long-acting nanoparticulate DNase-1 for effective suppression of SARS-CoV-2-mediated neutrophil activities and cytokine storm. Biomaterials. 2021;267:120389.
- Liu F, Li L, Xu M, et al. Prognostic value of interleukin-6, C-reactive protein, and procalcitonin in patients with COVID-19. J Clin Virol. 2020;127:104370.
- Tian W, Jiang W, Yao J, et al. Predictors of mortality in hospitalized COVID-19 patients: a systematic review and meta-analysis. J Med Virol. 2020;92(10):1875–1883.
- Cavalier E, Guiot J, Lechner K, et al. Circulating nucleosomes as potential markers to monitor COVID-19 disease progression. Front Mol Biosci. 2021;8:600881.
- Gómez RM, López Ortiz AO, Schattner M. Platelets and extracellular traps in infections. Platelets. 2021;32(3):305–313.
- Sung PS, Hsieh SL. C-type lectins and extracellular vesicles in virus-induced NETosis. J Biomed Sci. 2021;28(1):46.
- Njeim R, Azar WS, Fares AH, et al. NETosis contributes to the pathogenesis of diabetes and its complications. J Mol Endocrinol. 2020;65(4):R65–r76.
- Gavriilidis E, Antoniadou C, Chrysanthopoulou A, et al. Combined administration of inhaled DNase, baricitinib and tocilizumab as rescue treatment in COVID-19 patients with severe respiratory failure. Clin Immunol. 2022;238:109016.
- Kocak M, Ezazi Erdi S, and Jorba G, et al. Targeting autophagy in disease: established and new strategies. Autophagy. 2021;18(3):473–495.
- Calender A, Israel-Biet D, Valeyre D, et al. Modeling potential autophagy pathways in COVID-19 and sarcoidosis. Trends Immunol. 2020;41(10):856–859.
- Wang J, Ren X-R, Piao H, et al. Niclosamide-induced Wnt signaling inhibition in colorectal cancer is mediated by autophagy. Biochem J. 2019;476(3):535–546.
- Kindrachuk J, Ork B, Hart BJ, et al. Antiviral potential of ERK/MAPK and PI3K/AKT/mTOR signaling modulation for Middle East respiratory syndrome coronavirus infection as identified by temporal kinome analysis. Antimicrob Agents Chemother. 2015;59(2):1088–1099.
- Yang CC, Wu CJ, and Chien CY, et al. Green tea polyphenol catechins inhibit coronavirus replication and potentiate the adaptive immunity and Autophagy-dependent protective mechanism to improve acute lung injury in mice. Antioxidants (Basel). 2021;10(6):928.
- Guo X, Zhang M, Zhang X, et al. Porcine epidemic diarrhea virus induces autophagy to benefit its replication. Viruses. 2017;9(3):53.
- Egan DF, Chun MH, Vamos M, et al. Small molecule inhibition of the Autophagy kinase ULK1 and identification of ULK1 substrates. Mol Cell. 2015;59(2):285–297.
- Petherick KJ, Conway OJL, Mpamhanga C, et al. Pharmacological inhibition of ULK1 kinase blocks mammalian target of rapamycin (mTOR)-dependent autophagy. J Biol Chem. 2015;290(18):11376–11383.
- Liu P-F, Tsai K-L, Hsu C-J, et al. Drug repurposing screening identifies tioconazole as an ATG4 inhibitor that suppresses autophagy and sensitizes cancer cells to chemotherapy. Theranostics. 2018;8(3):830–845.
- Zhu H, Chen CZ, Sakamuru S, et al. Mining of high throughput screening database reveals AP-1 and autophagy pathways as potential targets for COVID-19 therapeutics. Sci Rep. 2021;11(1):6725.
- Dikic I, Elazar Z. Mechanism and medical implications of mammalian autophagy. Nat Rev Mol Cell Biol. 2018;19(6):349–364.
- Xue Z, Zhang Z, Liu H, et al. lincRNA-Cox2 regulates NLRP3 inflammasome and autophagy mediated neuroinflammation. Cell Death Differ. 2019;26(1):130–145.
- Bilezikian JP, Bikle D, Hewison M, et al. MECHANISMS IN ENDOCRINOLOGY: vitamin D and COVID-19. Eur J Endocrinol. 2020;183(5):R133–r147.