
ABSTRACT
Guanine-quadruplex structures (G4) are unusual nucleic acid conformations formed by guanine-rich DNA and RNA sequences and known to control gene expression mechanisms, from transcription to protein synthesis. So far, a number of molecules that recognize G4 have been developed for potential therapeutic applications in human pathologies, including cancer and infectious diseases. These molecules are called G4 ligands. When the biological effects of G4 ligands are studied, the analysis is often limited to nucleic acid targets. However, recent evidence indicates that G4 ligands may target other cellular components and compartments such as lysosomes and mitochondria. Here, we summarize our current knowledge of the regulation of lysosome by G4 ligands, underlying their potential functional impact on lysosome biology and autophagic flux, as well as on the transcriptional regulation of lysosomal genes. We outline the consequences of these effects on cell fate decisions and we systematically analyzed G4-prone sequences within the promoter of 435 lysosome-related genes. Finally, we propose some hypotheses about the mechanisms involved in the regulation of lysosomes by G4 ligands.
Introducing G-quadruplexes
Guanine-quadruplex structures (G4) are unusual nucleic acid conformations formed by guanine-rich DNA and RNA sequences (). These structures result from the stacking of two or more G-quartets, each composed of four coplanar guanines establishing a cyclic network of hydrogen bonds (). Although sharing the same elemental building block, G-quadruplexes are structurally diverse, displaying inter- or intra-molecular conformations, with different types of loops and potentially involving additional structural elements such as bulges or capping base pairs and triads. Depending on the relative orientation of the four strands, quadruplexes can be parallel (all strands running in the same direction), antiparallel (two in one direction, the others in the reverse orientation) or hybrid (3 + 1). These topologies differ in their overall shape, nature of the loops and accessibility of the terminal quartets and grooves (for a review please refer to [Citation1]). These polymorphic structures were first identified and intensively characterized in vitro with synthetic nucleic acid sequences, and direct evidence for G4 formation in cells came from studies using structure-specific antibodies [Citation2], in cell NMR [Citation3], and from analyses of genomic instability [Citation4]. Regarding their functional properties, G4 structures have been proposed to control a number of cellular processes, mostly related to their action at the DNA or RNA level, including telomere maintenance, DNA replication initiation and replication fork progression, transcription and translation [Citation5]. Hence, G4 play a pivotal role in maintaining normal cellular homeostasis through regulation of genomic stability as well as important physiological processes such as cell proliferation, differentiation, senescence and, in a few examples, development (for a review on the latter subject, please refer to [Citation6]). It is worth noting that the dynamics of formation and resolution of G-quadruplexes and their spatiotemporal localization are important in defining their cell type-specific biological effects. For example, G4-DNA landscapes differ among primary neurons, astrocytes and microglia and this variability impacts the susceptibility of cells to enter into senescence [Citation7]. Moreover, differential gene expression analyses of diverse cancerous and non-malignant cells highlight that the G4 folding state can determine the cell type-specific transcriptome [Citation8].
Figure 1. G4 structures and G4 ligands. (A) Presentation of a G-quartet, a planar cyclic arrangement of four guanines held together by Hoogsteen hydrogen bonds. Stacking of two or more quartets leads to the formation of a G-quadruplex. (B) Example of a structure composed of three stacked quartets, connected by three loops – two lateral and one “chain reversal” in this example, typical of a DNA quadruplex with a so called “hybrid” topology. RNA quadruplexes tend to be all-parallel, with chain-reversal loops (for a review on quadruplex structures and topologies, please refer to [Citation106]. (C) Presentation of the G-quadruplex ligands (G4L) cited in this review; these compounds belong to chemically-distinct families. T5 is a naphtalene diimide (NDI) derivative conjugated to a carbohydrate lactose, while 3a is a naphthalimide-benzotriazole conjugate. See for details. More examples of G4L can be found in the G4L database: https://www.g4ldb.com.
![Figure 1. G4 structures and G4 ligands. (A) Presentation of a G-quartet, a planar cyclic arrangement of four guanines held together by Hoogsteen hydrogen bonds. Stacking of two or more quartets leads to the formation of a G-quadruplex. (B) Example of a structure composed of three stacked quartets, connected by three loops – two lateral and one “chain reversal” in this example, typical of a DNA quadruplex with a so called “hybrid” topology. RNA quadruplexes tend to be all-parallel, with chain-reversal loops (for a review on quadruplex structures and topologies, please refer to [Citation106]. (C) Presentation of the G-quadruplex ligands (G4L) cited in this review; these compounds belong to chemically-distinct families. T5 is a naphtalene diimide (NDI) derivative conjugated to a carbohydrate lactose, while 3a is a naphthalimide-benzotriazole conjugate. See Table 1 for details. More examples of G4L can be found in the G4L database: https://www.g4ldb.com.](/cms/asset/f8491289-08ce-4c59-8873-fe11be8383da/kaup_a_2170071_f0001_oc.jpg)
Table 1. Effect of G-quadruplex ligands on autophagy.
The importance of G4 in anticancer strategies is being increasingly recognized due to the fact that G4-prone sequences may interfere with DNA replication and genomic stability and may be found at telomeres and within the promoters of several key oncogenes [Citation9]. Likewise, analysis of the regulatory genome regions of several viruses revealed the presence of G4-prone motifs in these regions arguing in favor of targeting G4 as an anti-viral strategy [Citation10–12]. Moreover, several G4 ligands elicited anti-proliferative and anti-replicative effects against cancer cells and viruses, respectively, highlighting once again their potential therapeutic benefits.
Bioinformatic analysis revealed that a high number of cellular DNA and RNA sequences are potentially able to adopt a quadruplex fold. Several algorithms have been proposed to predict G4 propensity [Citation13]. Depending on the method and parameters chosen, between 300,000 and 1 million potential G4 sites are predicted in the human genome. Interestingly, G4-prone motifs tend to be enriched at certain regions of the genome such as promoters or the first introns of mRNA [Citation14]. Depending on promoter definition (size window) and algorithm/parameters chosen, 50% or more of human genes have been reported to be potentially regulated by G4 motifs in their promoters [Citation15]. Furthermore, the latest nearly gapless “telomere-to-telomere” release of the human genome revealed an even higher number of candidate sequences, especially with respect to highly stable quadruplexes [Citation16]. The number of G4 actually formed within the genome at a given time is probably much lower and may depend on DNA sequence [Citation17], the phase of the cell cycle or on malignant transformation [Citation18]. The same applies to the RNA level since human cells have a robust machinery that globally unfolds RNA G-quadruplexes [Citation19].
G4 may be selectively recognized by small compounds called G-quadruplex ligands (G4Ls) and up to date thousands of G4Ls have now been reported [Citation20] (). Most of these compounds are cationic planar aromatic molecules, thus allowing favorable electrostatic interactions with a negatively-charged quadruplex, and proper stacking on a terminal G-quartet, which provides a convenient close-to-flat platform to interact with such ligands. There are interesting exceptions though. A negatively-charged porphyrin derivative such as NMM (N-methyl mesoporphyrin IX), a compound that becomes fluorescent when bound to G-quadruplexes (a so-called “light-up fluorescent probe” for G4) [Citation21] and non-planar steroid derivatives [Citation22] can also stabilize G4. On the contrary, G4L with many positive charges often exhibit limited specificity. These data suggest that a trade-off between affinity (often increased for G4L bearing multiple cationic groups) and selectivity (compounds that may bind to any negatively-charged biopolymer) needs to be explored [Citation23]. From a drug development perspective, the affinity and selectivity of each G4L should be defined to minimize off-target effects and toxicity. Some compounds such as PhenDC3 exclusively bind to G4, while others such as TMPyP4 recognize a broader range of nucleic acid structures [Citation24].
In summary, G4L are diverse and do not belong to a unique chemical family. It is important to consider that, while some compounds may exhibit higher affinity for one G4 topology [Citation25], the design of a ligand able to bind to a single G-quadruplex structure within the genome is currently elusive, meaning that multiple target sites are therefore expected to be bound by each G4L.
G4L were originally designed as telomerase inhibitors, as vertebrate telomeric repeats (TTAGGG)n are compatible with stable G4 formation. Indeed maintaining the telomeric DNA substrate into a “locked” folded conformation may prevent extension by telomerase [Citation26]. As telomerase activity is necessary for the sustained proliferation of cancer cells, its inhibition may have antineoplastic effects especially on tumors with short telomeres. However, telomeric effects observed with G4L may also be mediated by direct uncapping of the chromosomal extremities, and true enzymatic inhibition of telomerase is often overestimated [Citation27]. In addition, these compounds also recognize G4 motifs elsewhere in the genome. Autoradiography of metaphase spreads from cells cultured with a radiolabeled pyridine dicarboxamide G4L (called 360A) revealed that this ligand was bound to chromosome terminal regions, but also to interstitial sites [Citation28]. This indicates that such compounds may modulate the expression of numerous processes. At the transcriptional level, while most studies have been focused on genes transcribed by POLR2 (RNA polymerase II), some G4L reportedly affect the transcription of ribosomal genes [Citation29,Citation30]. Two G4L (quarfloxin and CX-5461) reached clinical trials for cancer treatment and were both reported to interfere with POLR1 (RNA polymerase I)-mediated DNA-to-RNA transcription [Citation31]. It is worth noting that some G4L elicit DNA damage responses and cell killing and have been shown to be particularly effective in cancer cells deficient in DNA repair genes [Citation32,Citation33].
Unfortunately, while showing interesting activity in cellulo [Citation34], G4L have often exhibited limited efficacy in in vivo disease models. There is therefore a critical need to better identify and characterize G4L cellular targets with the objective to discover new therapeutical strategies aimed to improve their efficacy in human diseases.
When the biological consequences of cell treatment with G4L are studied, the analysis is often limited to nucleic acid targets, the DNA damage response/repair, telomere dysfunction and gene expression. However, recent evidence revealed that, apart from the regulation of nucleus processes, G4L can target other cellular compartments such as mitochondria and lysosomes, suggesting that G4L may play more extensive roles in cell biology than previously appreciated. Here, we summarize the current knowledge of the relationships between lysosome and G4L underlying their potential effects on lysosome functions, lysosome biogenesis as well as transcriptional regulation of lysosomal genes. Meanwhile, we survey the impact of these regulations on autophagy, cellular senescence, cell cycle, cell death as well as immune responses. Finally, we present here our recent data in which we analyzed the propensity of lysosomal genes to be regulated by G4 formation using the G4-Hunter algorithm [Citation14,Citation35].
Lysosomes
Lysosomes are membrane-bound organelles that are well-known for their ability to degrade intracellular and extracellular material from the autophagic and endocytic or phagocytic trafficking pathways, respectively [Citation36,Citation37]. A large body of evidence has established that lysosomes can also operate as an intracellular signaling platform involved in cellular stress responses and in transcription programs to regulate cell proliferation and growth as well as nutrient sensing [Citation38]. The main sensor of this platform is MTOR (mechanistic target of rapamycin kinase) complex 1 (MTORC1) which, under nutrient and energy abundance promotes anabolism and cell growth while inhibiting catabolism and transcriptional regulation of lysosomal genes through the inhibition of autophagy and TFEB (transcription factor EB), respectively [Citation39,Citation40]. Here, we briefly introduce autophagy and TFEB, two key players in lysosomal functions and regulation that link lysosomes to the cellular stress response ().
Figure 2. A simplified model of the lysosome-autophagy-TFEB axis. Autophagy is a lysosomal process involved in the degradation and recycling of cellular components such as proteins, lipids and mitochondria. This process is orchestrated by several proteins known as ATG (autophagy related) proteins. Autophagy occurs through multi-step processes including 1) the initiation, formation and expansion of a double-membrane structure, known as a phagophore; 2) the phagophore then encloses cytoplasmic cargoes and seals to form a closed vesicle called an autophagosome; 3) the fusion of the complete autophagosome with the lysosome to form the autolysosome; 4) the degradation of sequestered cargoes by lysosomal hydrolases. As a result, new pools of energy-rich substrates and precursors for anabolic reactions are produced. In response to several stressors (e.g., starvation, DNA damage, lysosomal stress, etc.), autophagy is activated as a result of MTORC1 inactivation. MTORC1 also coordinates the transcription activity of TFEB. Active MTORC1 recruited to the lysosome promotes the phosphorylation of TFEB that leads to TFEB sequestration in the cytosol. Conversely, when MTORC1 is inactivated, TFEB is no longer phosphorylated, allowing its translocation from the cytosol to the nucleus. As a consequence, TFEB binds to the promoter region of several lysosomal and autophagy genes containing coordinated lysosomal expression and regulation (CLEAR) sequences and promotes their transcription.
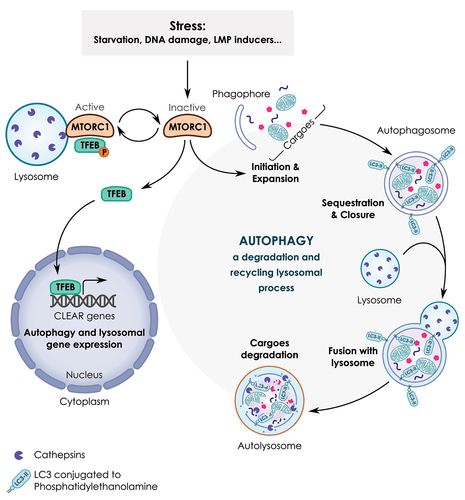
Macroautophagy/autophagy (herein simply referred to as “autophagy”) is a phylogenetically conserved catabolic process involved in the degradation and recycling of cellular components through the lysosomal pathway [Citation41–43]. Autophagy occurs through a multi-step process that requires the participation of several proteins known as ATG (autophagy related) proteins. During the initiation of autophagy, MTORC1 is inhibited leading to the stimulation of the ULK1 (unc-51 like autophagy activating kinase 1)-ULK2 complex and subsequent activation of the ATG proteins cascade. As a consequence, cellular cargoes are sequestered into a double membrane vesicle named autophagosome which then fuses with the lysosome to generate an autolysosome (). The sequestered material is then degraded by hydrolases in the lysosomal lumen, which allows the generation of energy-rich molecules and new pools of precursors for macromolecules synthesis and metabolic needs to ensure cell adaptation and survival [Citation44,Citation45]. Autophagy also promotes the selective removal of damaged organelles such as mitochondria and lysosomes (termed as mitophagy and lysophagy, respectively) and protein aggregates to preserve cell viability [Citation46,Citation47]. Selective autophagy is mediated by autophagic receptor proteins which recognize specific cargoes and deliver them to the autophagosome for subsequent degradation by lysosomal hydrolases [Citation48].
TFEB is a master regulator of autophagy/lysosomal genes that participate in the cellular stress response [Citation49,Citation50]. TFEB and TFE3 (transcription factor E3) belong to the microphthalmia (MIT/TFE) family of basic helix-loop-helix–leucine-zipper transcription factors [Citation49,Citation51] (). These proteins are structurally related and conserved through evolution; their tissue expression are highly regulated by alternative splicing and promoter usage, and their activity is mostly regulated by posttranslational modifications (for a recent review see Ref [Citation52]). Although the mechanisms regulating the activity of TFEB are well-documented, the regulation of its own transcription is still poorly understood. The human TFEB gene encodes a 2.4-kilobase (kb) mRNA transcript, starting with two non-coding exons followed by eight coding exons. Alternative transcripts [Citation53] result from the alternative inclusion of initial exons and are driven by different promoters [Citation54]. In response to starvation, the activation of TFEB leads to the induction of its own transcription, which represents a positive feedback loop to cope with stress [Citation55]. The subcellular localization and activity of TFEB are tightly controlled by MTORC1 [Citation56]. Under nutrient-rich conditions, the activation of MTORC1 on the lysosome promotes the phosphorylation of TFEB at serine residues 142 and 211 which results in TFEB retention in the cytosol [Citation57]. Conversely, inactivation of MTORC1 (for example during nutrient limitation or lysosomal stress) causes dephosphorylation of TFEB, allowing its translocation from the cytoplasm to the nucleus. As a result, TFEB binds to the promoter region of several lysosomal and autophagy genes containing coordinated lysosomal expression and regulation (CLEAR) elements (corresponding to a common 10-bases (GTCACGTGAC) E-box-like palindrome) [Citation58]. Examples of TFEB transcriptional target genes are: LAMP1 (lysosomal associated membrane protein 1) and LAMP2, the gene products of which stabilize the lysosomal membrane and interact with other cellular structures, genes encoding cathepsins that are implicated in degradation of intracellular and exogenous cellular material, and ATG genes (e.g., BECN1 [beclin 1] and GABARAP [GABA type A receptor-associated protein]) that are involved in different steps of the autophagy process [Citation59]).
Beyond serving as a signaling platform for cell stress response, the lysosome can also regulate cell death modalities as observed during lysosome membrane permeabilization (LMP) following chemical induction of lysosomal membrane instability [Citation60,Citation61]. Emerging evidence, however, suggest that several non-lethal functions of LMP can come into action during normal biological processes such as mitosis and cell adhesion [Citation62]. Lysosome membrane stability is regulated by several factors, such as the lipid composition of the lysosomal membrane (in particular the cholesterol/sphingolipid ratio), the proper function of proteins that operate as safeguards of lysosomal integrity including the class III phosphatidylinositol 3-kinase, HSPA/HSP70 (heat shock protein family A (Hsp70)), LAMP1 and LAMP2, as well as the redox status of lysosomal membrane [Citation60,Citation61]. At the molecular level, the induction of LMP leads to the activation of adaptive stress responses in order to repair (through the endosomal sorting complex required for transport, endosomal sorting complex required for transport (ESCRT) machinery), replace (via TFEB activation) and recycle damaged lysosomes (through lysophagy) [Citation61,Citation63]. If the induction of adaptive stress responses is unable to cope with prolonged lysosomal stress, the activation of LMP can result in the release of lysosomal hydrolases to the cytoplasm and subsequent cell death induction through both caspase-dependent and -independent mechanisms [Citation60,Citation61].
Some G4 ligands have a tropism for lysosomes or mitochondria
While quadruplex ligands usually reach the cell nucleus and modulate gene expression, there have been reports of G4L accumulating in cellular organelles as lysosome and mitochondria. The first evidence linking G4L to lysosomes came from the study of the G4 ligand 3,6-bis(1-methyl-4-vinylpyridium) carbazole diiodide (BMVC), a fluorescent probe displaying high sensitivity and binding preference to quadruplex DNA over non-G4 structures. BMVC is retained in the lysosomes of normal cells but escapes lysosomal retention in cancer cells to localize in the mitochondria or the nuclei of these cells [Citation64]. Structure-function analyses of a panel of BMVC derivatives revealed that hydrogen-bonding capacity drives lysosomal retention in normal cells, whereas, in cancer cells, lipophilicity governs the preferential localization of BMVC derivatives to the mitochondria, highlighting that the physicochemical properties of G4L including net charge, pKa value(s) and lipophilicity dictate their subcellular localization [Citation64]. Of note, drug-resistant cancer cells exhibit increased lysosomal BMVC retention as compared to drug-sensitive cancer cells, an effect that can be reversed by treatment with L-leucyl-L-leucine methyl ester, a lysosomotropic agent [Citation64]. In the same vein, Chang and colleagues showed that BMVC and its analog o-BMVC accumulate not only in the nuclei of cells but also in the lysosomal compartment [Citation65]. This accumulation of BMVC derivatives into lysosomes mirrors a process referred to as “lysosomal drug sequestration” that has been widely considered as a mechanism of resistance to some anti-cancer compounds [Citation66]. This process depends on cellular context and on physicochemical properties of the compound.
Another G4L that was found to accumulate into the lysosome is a ruthenium(II) polypyridyl complex (RPD) [Citation67]. Yu and colleagues showed that RPD enters HeLa cells through a non-endocytotic process that does not require an energy-dependent mechanism and then accumulates in lysosomes before escaping to end up in the nuclei. Moreover, they found that RPD induces apoptosis in HeLa cells through a mitochondrion-dependent pathway and inhibition of telomerase activity.
Besides the aforementioned G4L, we observed that agents belonging to a third family of G4L, the triarylpyridine derivatives exemplified by 20A, accumulate in the lysosome and causes the enlargement of this cellular compartment [Citation68].
A body of evidence showed that G4L can also accumulate in mitochondria. Here, we briefly present the literature regarding the potential roles of G4 in mitochondria, which have been covered before [Citation69]. The double-stranded circular human mitochondrial genome is rich in potential G4, and many of these sequences experimentally form quadruplexes under physiological conditions [Citation14]. The high density in G4-prone motifs comes from the asymmetry in strand composition of mitochondria DNA, with an enrichment of guanines on one strand (“GC skewness”). These quadruplexes may cause mitochondrial DNA deletions and contribute to the regulation of mitochondrial gene expression [Citation70–72]. In addition, a number of G4L have been found to localize to mitochondria (e.g., cyanine derivatives DODC and BMVC) [Citation73,Citation74].
Mitochondrial targeting G4L may also be useful to monitor mitophagy. A supramolecular FRET dye formed by two cyanine dyes is selectively located in the lysosomes of live cells and emits FRET fluorescence which gradually decreases in the presence of mitochondrial G4. This probe can therefore be applied to monitor the degradation of the mitochondrial G4 through the autophagy/lysosomal pathway [Citation75]. Similarly, the G4 ligand BYM, which was considered to be more selective for mitochondrial G4 than for other non G4 structures can be also used to track mitochondrial G4 in cells and monitor the dynamic process of mitophagy [Citation76].
Lysosomal uptake of exogenous G-quadruplexes
It has been reported that several G-rich DNA oligonucleotides (referred to as GROs) have the ability to accumulate in lysosomes. For example, Chang and colleagues studied the uptake and localization of exogenous G4-BMVC complex using guanine-rich (G-rich) sequences. They found that G-rich oligonucleotides harboring parallel G4 structures accumulate within the lysosome of lung cancer cells [Citation77]. Fluorescence imaging of exogenous G-rich DNA oligonucleotides showed that the exogenous parallel G4 structures are detected in the lysosomes of live cells in large quantities [Citation78]. Surprisingly, G-rich oligonucleotides adopting a different quadruplex conformation (nonparallel, as for the human telomeric motif), have a distinct behavior and are mostly detectable in mitochondria. G4-forming sequences exhibit enhanced cellular uptake as compared to non-G4 sequences, and this uptake may occur through the endosome/lysosome pathway [Citation79]. Once again, it is possible that the physicochemical properties of exogenous G4 may influence their selective enrichment in one particular cellular compartment rather than in others. The mechanism through which the structure of G4 can define its cellular localization deserves to be investigated in future studies. For a review on aptamer (including G4-forming sequences) uptake mechanism, please refer to [Citation80].
G4L can render cells hypersensitive to lysosome inhibitors
Evidence supports the idea that large lysosomes are more prone to lysosome leakage [Citation61]. In accordance with this assumption, we found that the combination of the G4L 20A with a lysosomotropic agent (either chloroquine or Lys 05) promotes a significantly more pronounced LMP as compared to 20A or chloroquine alone [Citation68]. The induction of LMP by 20A is accompanied by a significant induction of cell death, suggesting the contribution of lysosome leakage to cellular demise. Similar results were found with three other G4L from the triarylpyridine family, supporting the idea that this phenomenon is not specific to one single compound but can be observed with multiple G4L [Citation68]. The mechanisms through which G4L render cells more prone to lysosome leakage remain to be understood, as multiple mechanisms may hypothetically explain these effects, as follows: i) G4L-mediated lysosomal enlargement may be a prerequisite for the induction of LMP; ii) G4L may trigger destabilization of the lysosomal membrane through changes in its lipid composition (i.e., cholesterol or sphingolipid) by modulating the expression of key genes involved in lipid metabolism; iii) G4L might cause disruption of proteins that function as safeguards of lysosomal membrane integrity (e.g., the class III PtdIns3K and HSPA/HSP70); iv) G4L may trigger lipid peroxidation of the lysosomal membrane through modulating the redox metabolism of cells (via a transcriptional or non-transcriptional mechanism); and v) G4L may interact with the lysosomal membrane through hydrophobic or electrostatic interactions which could affect lysosomal membrane integrity.
Interestingly, these five processes rely on G4-transcriptional or non-transcriptional mechanisms that highlight additional or alternative modes of action of G4L that were not anticipated before.
Regulation of lysosomal genes by G4L
An expanding body of evidence indicates that lysosomal enlargement results in transcriptional induction of lysosomal genes, which represents a compensatory response for generating new lysosomes [Citation81,Citation82]. In line with these findings, we found that 20A-mediated lysosomal enlargement is associated with a significant enrichment of over 300 mRNAs relevant to the lysosomal pathway [Citation68,Citation83]. Interestingly, a much larger fraction (20.8%) of lysosomal genes were upregulated with a statistical cutoff of p < 0.001 than the proportion of lysosomal genes that were downregulated at a similar level of significance (8.5%). Among the genes and proteins whose expression is stimulated by 20A, one can find genes coding for lysosomal hydrolases and other enzymes (proteases: i.e. cathepsins; lipases: i.e. LIPA [lipase A, lysosomal acid type]; glycosidases: i.e. HEXA [hexosaminidase subunit alpha]; SMPD1 [shingomyelin phosphodiesterase 1]; ASAH1/acid ceramidase [N-acylsphingosine amidohydrolase 1]; lysosomal membrane proteins: LAMP1, LAMP2; genes coding for proteins involved in the transport of lysosomal enzymes: MGPR [mannose-6-phosphate receptor, cation dependent], clathrin, adaptor protein 3; as well as ATG genes; see below for detailed information). In the same vein, the gene expression data published by the Capranico laboratory revealed that non-cytotoxic pyridostatin (PDS) concentrations do not only promote the enrichment of immune-associated pathways but also induce the stimulation of the lysosome-related pathways in the MCF7 breast cancer cell line [Citation34,Citation84]. We performed a detailed analysis of this dataset and identified 398 genes associated to the lysosomal pathway whose expression levels were changed in response to PDS; among these genes 233 lysosomal genes were significantly (p < 0.001) upregulated and 164 genes were downregulated.
Altogether, the gene expression analyses indicate that a substantial number of lysosomal genes are altered in response to two distinct G4L, 20A and PDS. The mechanisms by which lysosome genes are regulated by 20A and PDS are not yet understood but may involve the participation of several regulatory pathways. Further transcriptomic studies are warranted to clarify the possible regulation of lysosomal genes by other G4L. In order to get a better understanding of the potential regulation of lysosomal genes by G4 sequences, we address the occurrence of G-quadruplex prone motifs in lysosomal and autophagy genes in the next chapter.
G-quadruplex prone motifs in lysosomal and autophagy genes
Transcriptomic analyses of lysosomal and autophagy genes (shown as supplementary data in [Citation68,Citation83] indicate that a number of such genes are modulated (and often upregulated) by G4L. To determine if this modulation could result from a general and direct transcriptional effect of G4L, we first determined whether the promoters of lysosomal and autophagy genes were enriched in G4 motifs as compared to all other human genes. A first study had already been performed on ATG genes (not only promoters) [Citation85], using a different algorithm called Quadruplex forming G-Rich Sequences (QGRS) mapper. For this, the authors used a very relaxed definition of promoters: 5 kb upstream of the transcription start site (TSS). Analyses revealed that most ATG genes contain several G4-prone motifs in their gene bodies, arguing for a possible role of G4 in the regulation of their expression. Note that these signatures are expected in view of the high proportion of human genes – at least 50% – exhibiting putative G4 motifs in their promoters.
To extend these results, we chose to reanalyze the promoters of 435 lysosomal genes listed in the Human Lysosome Gene Database (hLGDB) [Citation86] using the G4-Hunter algorithm previously developed by our group [Citation14,Citation35]. In , we compared G4 density in the promoters of lysosomal genes (including autophagy genes) versus all human genes. As G4 sequences reported to influence transcription have generally been found closer to the transcription start site (within 1 kb, and often much closer), we performed a search on a relatively short region (within 1 kb or 100 bp). As expected, we did find G4 motifs in the promoters of lysosomal and autophagy genes, but could not find evidence of any apparent specific enrichment in these promoters as compared to all human genes, suggesting that autophagy and lysosomal genes are not more susceptible to G4L than other genes (and 3B. This lack of a “strong G4 character” indicates that the lysosomal signature observed in response to some G4L is probably not the consequence of a direct “global” enrichment in G4 motifs in the lysosomal genes. However, we cannot definitely rule out the regulation of some lysosomal genes through G4-dependent mechanisms, and further transcriptomics and bioinformatics will be required to resolve this question.
Figure 3. G4 motifs in promoters. The density of G4 motifs found in the promoters of genes involved in lysosomal function (A) or autophagy genes (B), as compared to all human promoters (unpublished data). The search for G4 motifs is made with the G4-Hunter algorithm [Citation14,Citation35]. In panel (B), “TSS-1000” and “TSS+1000” refer to the 1-kb regions upstream and downstream of the Transcription Start Site (TSS), respectively. A restricted search in the immediate vicinity of the TSS (less than 100 bp away) is also provided, as we hypothesize that the most relevant G4 are actually those within the immediate vicinity of the TSS. No statistically significant difference is found between ATG genes (blue) and all genes (green) in any of the four categories. (C) Density of G4 motifs (as deduced by G4-Hunter, shown in red) in the vicinity of the TFEB promoter (note the presence of alternative transcription start sites). An example of a quadruplex sequence with a high-Hunter score very close to one TSS is provided.
![Figure 3. G4 motifs in promoters. The density of G4 motifs found in the promoters of genes involved in lysosomal function (A) or autophagy genes (B), as compared to all human promoters (unpublished data). The search for G4 motifs is made with the G4-Hunter algorithm [Citation14,Citation35]. In panel (B), “TSS-1000” and “TSS+1000” refer to the 1-kb regions upstream and downstream of the Transcription Start Site (TSS), respectively. A restricted search in the immediate vicinity of the TSS (less than 100 bp away) is also provided, as we hypothesize that the most relevant G4 are actually those within the immediate vicinity of the TSS. No statistically significant difference is found between ATG genes (blue) and all genes (green) in any of the four categories. (C) Density of G4 motifs (as deduced by G4-Hunter, shown in red) in the vicinity of the TFEB promoter (note the presence of alternative transcription start sites). An example of a quadruplex sequence with a high-Hunter score very close to one TSS is provided.](/cms/asset/91fb9742-f0ff-465c-a416-ffb3371f1946/kaup_a_2170071_f0003_oc.jpg)
As TFEB is a master regulator of lysosomal genes, we next explored whether G4-prone motifs may overlap the consensus DNA binding site of the TFEB transcription factor (the CLEAR element with a 10-bp consensus GTCACGTGAC) [Citation87]. This sequence is unlikely to overlap G4-prone motifs as it does not contain a single run of two or more consecutive guanines. We can therefore exclude a direct modulation of TFEB target genes by G4L via binding to CLEAR sequence in the target genes promoters.
Next, we examined the possible regulation of the transcription of the TFEB gene itself through a G4 binding-dependent mechanism. Based on the G4-Hunter tool, we found that the TFEB promoter gene contains G-rich motifs that are highly prone to G4 formation. To test whether G4L may have lysosomal effects via such a master gene regulator, we analyzed the TFEB gene in more details. Interestingly, the density of G4 motifs in the vicinity of TFEB transcription start sites (TSS) is extremely high () and some of these TSS are very close to strong G4-forming sequences in the TFEB gene (e.g., the distance between one TSS and the candidate sequence provided in is only 2 base pairs). The presence of multiple G4 sites close to the TSS may explain how G4L modulates the expression of the TFEB gene which may subsequently drives the specific transcription of lysosomal genes.
To our knowledge, apart from the transcriptomics analyses of lysosomal and autophagy genes found in [Citation34,Citation68,Citation83], there are no data in the literature on TFEB gene expression regulation by G4 structures or G4L. This is an interesting research area that deserves further investigation to better understand the mechanism underlying TFEB gene expression and its biological significance in various pathophysiological settings.
Regulation of autophagy by G4L
The first evidence linking G4L to the autophagy process came from the studies of the Zhu laboratory working on the G4L SYUIQ-5, a cryptolepine derivative [Citation88] (see). The authors found that SYUIQ-5 triggers the accumulation of MAP1LC3/LC3 (microtubule associated protein 1 light chain 3) puncta and the formation of telomeric γ-H2AX (H2A.X variant histone) family member foci in cervical and nasopharyngeal cancer cells. Inhibition of ATM (Ataxia Telangiectasa Mutated) protein kinase, a key component of the DDR, attenuates the accumulation of both γ-H2AX foci and MAP1LC3/LC3 puncta, supporting the idea that ATM contributes to autophagy induction and the DNA damage response (DDR) measured by the quantification of γ-H2AX foci. In a subsequent study, the same group showed that SYUIQ-5 concomitantly promotes nuclear translocation of the FOXO3 (forkhead box O3) transcription factor and upregulation of mRNA and protein levels of two autophagy-relevant proteins, MAP1LC3 and BNIP3 (BCL2 interacting protein 3) [Citation89]. Knockdown of BNIP3 expression attenuates the accumulation of LC3-II, suggesting the contribution of BNIP3 to SYUIQ-5-induced autophagy. These results support the possibility that FOXO3 mediates autophagy stimulation in response to SYUIQ-5. Future studies must elucidate the precise mechanism by which FOXO3 and ATM cooperate to stimulate autophagy in response to SYUIQ-5.
One important piece of evidence linking G4L to autophagy came from the study of the Zaffaroni laboratory that investigated the effect of an anthracene derivative called Ant1,5 [Citation90]. The authors found that short-term exposure of melanoma cells to Ant1,5 causes cell growth arrest and induces telomere dysfunction without any evidence of senescence or apoptosis. Apparently, Ant1,5 treatment induces autophagy occurs as a consequence of DNA damage induction due to telomere uncapping. The induction of autophagy was found to be dependent on CDKN1A (cyclin dependent kinase inhibitor 1A) but occurred independently of TP53 (tumor protein 53) and MTOR activity.
We also investigated the effect of 20A on autophagy and explored how this regulation may affect cell fate in cancer cells [Citation91]. From the transcriptomic and Kyoto Encyclopedia of Genes and Genomes (KEGG) database analyses, we first found that 20A treatment of HeLa cells elicits a significant functional enrichment of genes related to the autophagy pathway. These genes can be classified in three categories as follows: i) genes encoding for the components of the autophagy machinery (e.g., BECN1, PIK3R4 [phosphoinositide 3-kinase regulatory subunit 4], ATG5 [autophagy related 5], ATG12, ATG16L1, ATG3, MAP1LC3); ii) genes coding for autophagy receptors (e.g., SQSTM1 [sequestosome 1], OPTN [optineurin], CALCOCO2 [calcium binding and coiled-coil domain 2], NBR1 [NBR1 autophagy cargo receptor]); iii) genes encoding for autophagy regulators (e.g., PRKAA [protein kinase AMP-activated alpha catalytic subunit], TSC1 [TSC complex subunit 1] and TSC2, RHEB [Ras homolog, mTORC1 binding]) and several elements of the MTORC1 complex. Consistent with the transcriptomic data, we found that treatment of HeLa cells with 20A elicits autophagy induction associated with the activation of PRKAA and inhibition of MTORC1 activity. The induction of autophagy occurred as a consequence of global DNA damage (but not specific telomeric damage) that triggered the activation of ATM . Pharmacological and genetic inhibition of either autophagy or ATM causes a significant inhibition of 20A-induced senescence with a concomitant activation of apoptotic cell death [Citation83,Citation91].
In addition to 20A, enrichment of autophagy-related Gene Ontology (GO) terms was observed in MCF-7 breast cancer cells treated with cytostatic concentrations of PDS [Citation34]. Detailed analysis of gene expression datasets showed that the mRNA expression levels of two autophagy receptors OPTN and SQSTM1, as well as TFEB, were significantly upregulated in response to PDS [Citation34]. Such upregulations have been also observed with 20A, suggesting the existence of overlapping regulatory mechanisms for two distinct G4L, 20A and PDS. Along with the activation of autophagy, PDS can trigger innate gene activation through a mechanism that involves the cGAS (cyclic GMP-AMP synthase)-STING1 (stimulator of interferon response cGAMP interactor 1)-IRF3 (interferon regulatory factor 3) pathway [Citation34]. Further studies must elucidate the link between stimulation of autophagy and innate gene activation in response to PDS.
In another study from the Zhang laboratory, the effect of a phenanthroline G4L derivative (13d) on the regulation of autophagy was investigated [Citation92]. The authors found that 13d treatment of gastric cancer cells results in autophagy activation secondary to DNA damage mediated by telomere dysfunction.
Li et al. explored the effect of the G4L CX-5461 on autophagy [Citation93]. This compound, also known as pidnarulex, was initially reported to inhibit POLR, but its real molecular target may be topoisomerase 2 at transcribed regions containing G4 [Citation94,Citation95]. Li et al showed that CX-5461 induces G2 cell cycle arrest and accumulation of LC3-II form in both U2OS cells and MNNG cells which is associated with MTOR inhibition and PRKAA activation. CX-5461 induces autophagy through a TP53-dependent mechanism and exerts synergistic anticancer effects when combined with doxorubicin. CX5461 was also shown to synergize with radiotherapy against cervical cancer cells, as reported by Ismael et al [Citation96]. Inhibition of POLR1 by either CX5461 or PICT overexpression induces pro-death autophagy through mechanisms that are still elusive [Citation97]. In line with these results, the Pfister laboratory recently reported that induction of nucleolar stress triggers expression of key autophagy regulators, and that of POLR1 inhibition by a G4L, CX-5461, correlated with increased expression levels of two key autophagy genes, ATG7 and ATG16L1 [Citation98]. Naphthalene-diimides belong to another G4 ligand family that was shown to target G-quadruplexes in ribosomal DNA [Citation99]. This action induces a rapid inhibition of POLR1 that leads to autophagy stimulation associated to cell death. The lead compound T5 mediates potent and selective antitumor effects against colorectal cancer cells but minimal toxicity on non-malignant cells, suggesting that T5 could be exploited for cancer treatment in colorectal cancer patients. Altogether, these findings corroborate other reports revealing the induction of autophagy under nucleolar stress caused by POLR1 inhibition [Citation31].
Recently, the Zhang laboratory investigated the potential anticancer activity of a series of novel G4L from the naphthalimide-benzotriazole series (1a–3c). They found that the lead compound 3a stabilized G4 structures in the gene coding for BCL2 (BCL2 apoptosis regulator) and induced cell death with features of apoptosis and autophagy [Citation100].
In sharp contrast with the above studies which reported the activation of autophagy by G4Ls, two studies from the Tsvetkov lab [Citation85,Citation101], showed that two distinct G4Ls, pyridostatin (PDS) and BRACO19, suppressed autophagy in post-mitotic neurons. The proposed mechanism for the inhibition of autophagy by PDS involves the downregulation of the key autophagy gene, ATG7, through stabilization of DNA-G4 structures present within this gene. In vitro assays corroborated that a putative G4-DNA forming sequence identified in the ATG7 gene folded into a stable G4 structure which can be recognized by PDS. Further studies are required to determine whether inhibition of autophagy caused by PDS in neurons relies solely on ATG7 gene downregulation or rather involves other yet unidentified regulatory mechanisms.
The inhibition of autophagy in neurons reported for PDS and BRACO19 differs from the effects observed for other G4L in cancer cells. One explanation would be the distinct effects of G4 on autophagy regulation in neurons versus cancer cells. In favor of this conjecture, it has been proposed that G4 folding state can regulate cell type-specific biological effects [Citation8]. Moreover, in particular regarding PDS, different experimental conditions (concentrations, duration of treatment) were used in neurons [Citation85] and cancer cells [Citation34], which may explain the variability of autophagy responses. Such differential regulation of autophagy has been also observed in neurons versus astrocytes in the context of metabolic stress [Citation7]. Future investigations must determine whether the regulation of autophagy by PDS (as well as other G4L) is dependent on cell type, the nature of the G4L, as well as on the concentration and duration of treatment.
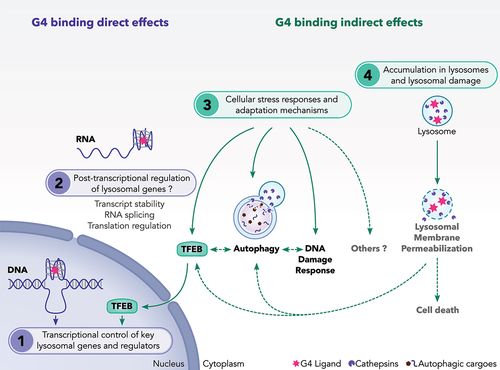
Taken together, these results indicate that G4L have the ability to regulate autophagy through several interconnected routes. We suggest that G4L regulate autophagy possibility through direct modulation of autophagy genes or through indirect effects caused by the activation of cellular stress responses (). In fact, bioinformatic evidence supports the possibility that G4L may directly regulate autophagy as well as their regulators (such as TFEB) through interaction with G4-DNA or G4-RNA sequences identified in autophagy-regulatory genes, as proposed for ATG7 and TFEB genes (see chapter”G-quadruplex prone motifs in lysosomal and autophagy genes” and ). Alternatively, autophagy can be influenced by G4L-mediated cellular stress responses resulting from nuclear and nucleolar stresses as well a rewiring of the transcriptome (). In this sense, several G4L exhibit MTORC1 inhibition () which is a critical and early event involved in the activation of adaptive stress responses such as autophagy and TFEB. It is worth noting that autophagy and TFEB may also be activated in response to those G4L that trigger LMP induction, although this possibility has not yet been explored in the literature . The induction of global or telomeric DNA damage represents another mechanism by which G4L may induce autophagy through activation of stress responses (). In turn, autophagy may regulate DNA damage induced by G4L by regulating the DNA damage response as seen for 20A (). Further studies are required to determine the exact molecular mechanisms underlying the direct and indirect effects of G4L on autophagy.
Figure 4. A proposed model showing the mechanisms of action of G4 ligands on lysosomes. We suggest that G4 ligands regulate the lysosomal pathway through multiple non exclusive mechanisms. These include: (1) Direct interaction of G4L with DNA-G4 motifs presented in the promoter of some lysosomal genes. G4L may also modulate the gene expression of TFEB transcription factor, a master regulator of lysosomal genes. (2) Direct interaction of G4L with G4 presented in RNAs relevant for autophagy and lysosomal function. So far, this regulation has not been investigated for this class of genes. (3) Activation of adaptive stress pathways such as autophagy, TFEB and DNA damage response (DDR) as a consequence of cellular stresses induced by G4L. (4) Induction of lysosomal membrane permeabilization as a consequence of the sequestration of G4L into the lysosomes. During LMP, adaptive stress responses including autophagy and TFEB are activated to cope with damaged lysosomes and to ensure cell survival. If these adaptive stress responses are unable to overcome the lysosomal stress, the LMP induction can result in cell death.
The role of autophagy in cell fate decisions upon G4L treatment
Depending on the precise G4L and cell type, the induction of autophagy can result in either a cytoprotective or a cytotoxic response. For example, inhibition of autophagy by a pharmacological inhibitor or through genetic depletion of ATG5 enhanced the cytotoxicity of Ant1,5 in melanoma cells, suggesting a cytoprotective role for autophagy in this context [Citation90]. Suppression of autophagy by deletion of ATG5 and ATG7 inhibits senescence induced by 20A in HeLa cells and instead promotes apoptosis [Citation83]. Moreover, knocking down the autophagy-related gene ATG5 enhances apoptosis induced by 13d in gastric cancer cells [Citation92]. This cytoprotective effect of autophagy may involve a variety of mechanisms including: i) selective degradation of damaged cellular components (lysosomes, mitochondria, and macromolecules); ii) selective recruitment of cell death regulatory components to autophagosomes for their degradation; iii) direct interaction of the autophagic proteins with the signaling components of cell death pathways; iv) sustained activation of cell metabolism as a consequence of the production of new nutrients and energy sources [Citation44].
Contrasting with this protective effect of autophagy, autophagy was shown to act as a pro-death mechanism in response to G4L, SYUIQ-5 [Citation88]. In fact, shRNA-mediated knockdown of ATG5 expression attenuates the cytotoxicity of SYUIQ-5. Similarly, pharmacological inhibition of autophagy by 3-methyladenine markedly reduces cell death induced by CX-5461 in osteosarcoma cell lines [Citation93], suggesting that autophagy contributes to cell death under these conditions through yet-to-be-unveiled mechanisms.
It is worth noting that some ATG proteins are involved in the control of cell death processes through an autophagy-independent mechanism, as reported for ATG5 and ATG12 [Citation102–104]. Hence, the possibility of such an autophagy-independent cell death modulation should be considered when studying the cytotoxic effects of G4L.
Beyond regulation of cell death, G4L may impact the aging process. In line with this assumption, PDS was found to inhibit autophagy in neurons and cause memory deficits in mice [Citation85,Citation101]. Further studies are required to determine the cause-effect relationship between autophagy modulation and aging in response to G4L. The finding that PDS can cause an aging phenotype in the mouse brains highlights the possible undesirable side effects of this ligand on normal cells. Thus, caution should be taken to define the appropriate doses and duration of treatment when PDS – or another G4L – is used for therapeutic purposes to avoid undesirable side effects on normal cells.
Collectively, the regulation of autophagy by G4L can result in different cellular outcomes that may be related to the nature of the compound, the concentration and duration of treatment, the affected cell type, and the specific signaling pathways activated by the G4L. An extensive analysis of the role of ATG proteins involved in different steps of autophagy should help to define the mechanisms and the consequences of autophagy regulation in response to a given G4L.
Conclusion and perspectives
The available evidence indicates that G4L from different families have the ability to control the lysosomal pathway in several distinct cell types. We propose that these regulations occur through several interconnected mechanisms that involve either direct effects through interaction with G4 motifs or indirect effects linked to activation of adaptive stress pathway, as summarized in . These include:
Direct interaction of G4L with DNA-G4 motifs present in the promoter of lysosomal genes and regulators. G4L may modulate the mRNA expression of the TFEB transcription factor, which is a master inducer of lysosomal genes.
Direct interaction of G4L with RNA-G4 motifs, as stable G4 formation is also possible for G-rich RNA motifs. RNA quadruplexes can actually regulate any gene expression step, from transcription to protein synthesis [Citation105] (i.e. transcript stability, RNA splicing, translation). So far, this regulation has not been investigated for autophagy/lysosome-related genes.
Activation of adaptive stress pathways (e.g., autophagy, TFEB and DNA damage responses) as a consequence of G4L-induced cellular stress responses possibly due to changes in transcriptional activity and/or induction of nuclear or nucleolar stresses.
Induction of LMP as a consequence of the sequestration of G4L within the lysosomes.
Interestingly, the regulation of lysosomes by G4L can result in different cellular outcomes that may be related to the chemical nature of the G4L, the concentration and duration of treatment and the intrinsic characteristics of the cell type. All of the G4L shown to act on autophagy or lysosomal pathways and described in are planar and positively charged at physiological pH: this is the case for most, but not all G4L, as a few of them (among over 3,000) are neutral (e.g., telomestatin) or even negatively charged (e.g., NMM). It will be interesting to test if such molecules induce the same effects.
On the one hand, the stimulation of the lysosome/autophagy pathway by G4L may result in a cytoprotective effect, implying that targeting lysosomes with specific inhibitors (such as chloroquine or hydroxychloroquine) would improve the efficacity of G4L against cancer cells and prevent resistance to treatment ( and [Citation68]). Such combinatorial drug strategy has already been used in several clinical trials with encouraging results[Citation104] On the other hand, the stimulation of the lysosomal pathway can contribute to cell death as seen for some G4L (). Thus, inhibiting or stimulating the lysosomal pathway in combination with G4L treatments can impact cell fate decisions, and both strategies could be of interest depending on the context and the specific disease. More work needs to be done to evaluate how lysosomes can be targeted to optimize the clinical application of G4L.
As for other DNA damaging agents that are already used in the clinic, prolonged treatment with G4L may result in undesirable side effects on normal cells such as DNA damage, mutation, cell senescence and accelerated aging [Citation33]. Further studies will have to define the doses and the duration of G4L treatments that provide therapeutic benefit, yet minimize side effects on normal cells.
We are confident that this review will stimulate further studies on the complex mechanisms through which G4L affect lysosomal biogenesis and function, hoping that such an endeavor will not only yield cognitive insights but might reveal druggable, disease-relevant pathways.
List of abbreviations
ATG: autophagy related
ATM: Ataxia Telangiectasa Mutated
BMVC: 3,6-bis(1-methyl-4-vinylpyridium) carbazole diiodide
BNIP3: BCL2 interacting protein 3
BRACO 19: N,N′-(9-(4-(dimethylamino)phenylamino)acridine-3,6-diyl)bis(3-(pyrrolidin-1-yl)propanamide)
DDR: DNA damage response
PtdIns3K: class III phosphatidylinositol 3-kinase
FOXO3: forhead box O3
γ-H2AX: gamma H2A.X variant histone
G4: G quadruplex structures that can be formed both with DNA (G4-DNA) or RNA (G4-RNA)
G4L: G-quadruplex ligand
GRO: G-rich DNA oligonucleotide
hLGDB database: human lysosome gene database
kb: kilobase pair
LAMP: lysosomal associated membrane protein
LMP: lysosome membrane permeabilization
MAP1LC3: microtubule associated protein 1 light chain 3
MTOR: mechanistic target of rapamycin kinase
PDS: pyridostatin
POLR: RNA polymerase
PRKAA: protein kinase AMP-activated alpha catalytic subunit
TFEB: transcription factor EB
TFE3: transcription factor E3
TSS: transcription start site
Acknowledgments
We thank Jean-Christophe Andrau, Anne Cucchiarini, Thomas Mardivirin and Zackie Aktary for helpful discussions.
Disclosure statement
GK has been holding research contracts with Daiichi Sankyo, Eleor, Kaleido, Lytix Pharma, PharmaMar, Osasuna Therapeutics, Samsara Therapeutics, Sanofi, Tollys, Vascage and Vasculox/Tioma. GK has been consulting for Reithera. GK is on the Board of Directors of the Bristol Myers Squibb Foundation France. GK is a scientific co-founder of everImmune, Osasuna Therapeutics, Samsara Therapeutics and Therafast Bio. GK, JR, M.D-M and JLM are the inventors of patents covering therapeutic targeting of aging, cancer, cystic fibrosis and metabolic disorders. None of these activities is linked to the current manuscript.
Additional information
Funding
References
- Phan AT. Human telomeric G-quadruplex: structures of DNA and RNA sequences. FEBS J. 2010;277:1107–1117.
- Biffi G, Tannahill D, McCafferty J, et al. Quantitative visualization of DNA G-quadruplex structures in human cells. Nat Chem. 2013;5:182–186.
- Salgado GF, Cazenave C, Kerkour A, et al. G-quadruplex DNA and ligand interaction in living cells using NMR spectroscopy. Chem Sci. 2015;6:3314–3320.
- Ribeyre C, Lopes J, Boulé J-B, et al. The yeast Pif1 helicase prevents genomic instability caused by G-quadruplex-forming CEB1 sequences in vivo. PLoS Genet. 2009;5:e1000475.
- Varshney D, Spiegel J, Zyner K, et al. The regulation and functions of DNA and RNA G-quadruplexes. Nat Rev Mol Cell Biol. 2020;21:459–474.
- Armas P, Calcaterra NB. G-quadruplex in animal development: contribution to gene expression and genomic heterogeneity. Mech Dev. 2018;154:64–72.
- Tabor N, Ngwa C, Mitteaux J, et al. Differential responses of neurons, astrocytes, and microglia to G-quadruplex stabilization. Aging (Albany NY). 2021;13:15917–15941.
- Lago S, Nadai M, Cernilogar FM, et al. Promoter G-quadruplexes and transcription factors cooperate to shape the cell type-specific transcriptome. Nat Commun. 2021;12:3885.
- Carvalho J, Mergny J-L, Salgado GF, et al. Friend or Foe: the role of the g-quartet in anticancer strategies. Trends Mol Med. 2020;26:848–861.
- Perrone R, Nadai M, Frasson I, et al. A dynamic G-quadruplex region regulates the HIV-1 long terminal repeat promoter. J Med Chem. 2013;56:6521–6530.
- Amrane S, Kerkour A, Bedrat A, et al. Topology of a DNA G-quadruplex structure formed in the HIV-1 promoter: a potential target for anti-HIV drug development. J Am Chem Soc. 2014;136:5249–5252.
- Abiri A, Lavigne M, Rezaei M, Nikzad S, Zare P, Mergny J-L, Rahimi H-R. Unlocking. G-Quadruplexes as antiviral targets. Pharmacol Rev. 2021;73:897–923.
- Puig Lombardi E, Londoño-Vallejo A. A guide to computational methods for G-quadruplex prediction. Nucleic Acids Res. 2020;48:1–15.
- Bedrat A, Lacroix L, Mergny J-L. Re-evaluation of G-quadruplex propensity with G4Hunter. Nucleic Acids Res. 2016;44:1746–1759.
- Brázda V, Bartas M, Bowater RP. Evolution of diverse strategies for promoter regulation. Trends Genet. 2021;37:730–744.
- Bohálová N, Mergny J-L BV. Novel G-quadruplex prone sequences emerge in the complete assembly of the human X chromosome. Biochimie. 2021;191:87–90.
- Piazza A, Adrian M, Samazan F, et al. Short loop length and high thermal stability determine genomic instability induced by G-quadruplex-forming minisatellites. EMBO J. 2015;34:1718–1734.
- Biffi G, Tannahill D, Miller J, et al. Elevated levels of G-quadruplex formation in human stomach and liver cancer tissues. PLoS One. 2014;9:e102711.
- Guo JU, DP B. RNA G-quadruplexes are globally unfolded in eukaryotic cells and depleted in bacteria. Science. 2016;353:aaf5371.
- Wang W, Zhou J, Shi J, et al. T-cell leukemia virus type 1 Tax-deregulated autophagy pathway and c-FLIP expression contribute to resistance against death receptor-mediated apoptosis. J Virol. 2014;88:2786–2798.
- Yett A, Lin LY, Beseiso D, et al. N-methyl mesoporphyrin IX as a highly selective light-up probe for G-quadruplex DNA. J Porphyr Phthalocyanines. 2019;23:1195–1215.
- Brassart B, Gomez D, De Cian A, et al. A new steroid derivative stabilizes g-quadruplexes and induces telomere uncapping in human tumor cells. Mol Pharmacol. 2007;72:631–640.
- Zuffo M, Guédin A, Leriche E-D, et al. More is not always better: finding the right trade-off between affinity and selectivity of a G-quadruplex ligand. Nucleic Acids Res. 2018;46:e115.
- Luo Y, Granzhan A, Verga D, et al. A fluorescence melting competition assay for studying G4 structures in vitro. Biopolymers. 2021;112:e23415.
- Hamon F, Largy E, Guédin-Beaurepaire A, et al. An acyclic oligoheteroaryle that discriminates strongly between diverse G-quadruplex topologies. Angew Chem Int Ed Engl. 2011;50:8745–8749.
- Sun D, Thompson B, Cathers BE, et al. Inhibition of human telomerase by a G-quadruplex-interactive compound. J Med Chem. 1997;40:2113–2116.
- De Cian A, Cristofari G, Reichenbach P, et al. Reevaluation of telomerase inhibition by quadruplex ligands and their mechanisms of action. Proc Natl Acad Sci U S A. 2007;104:17347–17352.
- Granotier C, Pennarun G, Riou L, et al. Preferential binding of a G-quadruplex ligand to human chromosome ends. Nucleic Acids Res. 2005;33:4182–4190.
- Drygin D, Siddiqui-Jain A, O’Brien S, et al. Anticancer activity of CX-3543: a direct inhibitor of rRNA biogenesis. Cancer Res. 2009;69:7653–7661.
- Drygin D, Lin A, Bliesath J, et al. Targeting RNA polymerase I with an oral small molecule CX-5461 inhibits ribosomal RNA synthesis and solid tumor growth. Cancer Res. 2011;71:1418–1430.
- Ferreira R, Schneekloth JS, Panov KI, et al. Targeting the RNA Polymerase I Transcription for Cancer Therapy Comes of Age. Cells. 2020;9:E266.
- Zimmer J, Tacconi EMC, Folio C, et al. Targeting BRCA1 and BRCA2 Deficiencies with G-Quadruplex-Interacting Compounds. Mol Cell. 2016;61:449–460.
- De Magis A, Manzo SG, Russo M, et al. DNA damage and genome instability by G-quadruplex ligands are mediated by R loops in human cancer cells. Proceedings of the National Academy of Sciences, USA. 2019; 116:816–825.
- Miglietta G, Russo M, Duardo RC, et al. G-quadruplex binders as cytostatic modulators of innate immune genes in cancer cells. Nucleic Acids Res. 2021;49:6673–6686.
- Brázda V, Kolomazník J, Lýsek J, et al. G4Hunter web application: a web server for G-quadruplex prediction. Bioinformatics. 2019;35:3493–3495.
- Saftig P, Puertollano R. How lysosomes sense, integrate, and cope with stress. Trends Biochem Sci. 2021;46:97–112.
- Lawrence RE, Zoncu R. The lysosome as a cellular centre for signalling, metabolism and quality control. Nat Cell Biol. 2019;21:133–142.
- Inpanathan S, Botelho RJ. The lysosome signaling platform: adapting with the times. Front Cell Dev Biol. 2019;7:113.
- Ben-Sahra I, Manning BD. mTORC1 signaling and the metabolic control of cell growth. Curr Opin Cell Biol. 2017;45:72–82.
- Liu GY, Sabatini DM. mTOR at the nexus of nutrition, growth, ageing and disease. Nat Rev Mol Cell Biol. 2020;21:183–203.
- Parzych KR, Klionsky DJ. An overview of autophagy: morphology, mechanism, and regulation. Antioxid Redox Signal. 2014;20:460–473.
- Galluzzi L, Baehrecke EH, Ballabio A, et al. Molecular definitions of autophagy and related processes. EMBO J. 2017;36:1811–1836.
- Levine B, Kroemer G. Biological functions of autophagy genes: a disease perspective. Cell. 2019;176:11–42.
- Kroemer G, Mariño G, Levine B. Autophagy and the Integrated Stress Response. Mol Cell. 2010;40:280–293.
- White E, Lattime EC, Guo JY. Autophagy Regulates Stress Responses, Metabolism, and Anticancer Immunity. Trends Cancer. 2021;7:778–789.
- Pohl C, Dikic I. Cellular quality control by the ubiquitin-proteasome system and autophagy. Science. 2019;366:818–822.
- Faruk MO, Ichimura Y, Komatsu M. Selective autophagy. Cancer Sci. 2021;112:3972–3978.
- Johansen T, Selective Autophagy: LT. ATG8 family proteins, LIR motifs and cargo receptors. J Mol Biol. 2020;432:80–103.
- Napolitano G, Ballabio A. TFEB at a glance. J Cell Sci. 2016;129:2475–2481.
- Raben N, Puertollano R. TFEB and TFE3: linking lysosomes to cellular adaptation to stress. Annu Rev Cell Dev Biol. 2016;32:255–278.
- Slade L, Pulinilkunnil T. The MiTF/TFE family of transcription factors: master regulators of organelle signaling, metabolism, and stress adaptation. Mol Cancer Res. 2017;15:1637–1643.
- La Spina M, Contreras PS, Rissone A, et al. MiT/TFE family of transcription factors: an evolutionary perspective. Front Cell Dev Biol. 2020;8:609683.
- Kuiper RP, Schepens M, Thijssen J, et al. Regulation of the MiTF/TFE bHLH-LZ transcription factors through restricted spatial expression and alternative splicing of functional domains. Nucleic Acids Res. 2004;32:2315–2322.
- Pérez-Roca L, Prada-Dacasa P, Segú-Vergés C, et al. Glucocerebrosidase regulators SCARB2 and TFEB are up-regulated in Lewy body disease brain. Neurosci Lett. 2019;706:164–168.
- Settembre C, Fraldi A, Medina DL, et al. Signals for the lysosome: a control center for cellular clearance and energy metabolism. Nat Rev Mol Cell Biol. 2013;14:283–296.
- Martina JA, Chen Y, Gucek M, et al. MTORC1 functions as a transcriptional regulator of autophagy by preventing nuclear transport of TFEB. Autophagy. 2012;8:903–914.
- Settembre C, Zoncu R, Medina DL, et al. A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. EMBO J. 2012;31:1095–1108.
- Settembre C, Medina DLTFEB. and the CLEAR network. Methods Cell Biol. 2015;126:45–62.
- Feng Y, He D, Yao Z, et al. The machinery of macroautophagy. Cell Res. 2014;24:24–41.
- Aits S, Jäättelä M. Lysosomal cell death at a glance. J Cell Sci. 2013;126:1905–1912.
- Wang F, Gómez-Sintes R, Boya P. Lysosomal membrane permeabilization and cell death. Traffic. 2018;19:918–931.
- Stahl-Meyer J, Stahl-Meyer K, Jäättelä M. Control of mitosis, inflammation, and cell motility by limited leakage of lysosomes. Curr Opin Cell Biol. 2021;71:29–37.
- Papadopoulos C, Kravic B, Meyer H. Repair or Lysophagy: dealing with Damaged Lysosomes. J Mol Biol. 2020;432:231–239.
- Kang -C-C, Huang W-C, Kouh C-W, et al. Chemical principles for the design of a novel fluorescent probe with high cancer-targeting selectivity and sensitivity. Integr Biol (Camb). 2013;5:1217–1228.
- Tse T-Y, Chang C-C, Lin J-J, Chang T-C. A Fluorescent Anti-Cancer. Agent, 3,6-bis(1-methyl-4-vinylpyridinium) carbazole diiodide, stains G-Quadruplexes in cells and inhibits tumor growth. Curr Top Med Chem. 2015;15:1964–1970.
- Zhitomirsky B, Assaraf YG. Lysosomes as mediators of drug resistance in cancer. Drug Resist Updat. 2016;24:23–33.
- Yu Q, Liu Y, Xu L, et al. polypyridyl complexes: cellular uptake, cell image and apoptosis of HeLa cancer cells induced by double targets. Eur J Med Chem. 2014;82:82–95.
- Beauvarlet J, Nath Das R, Alvarez-Valadez K, et al. Triarylpyridine compounds and chloroquine act in concert to trigger lysosomal membrane permeabilization and cell death in cancer cells. Cancers (Basel). 2020;12:E1621.
- Falabella M, Fernandez RJ, Johnson FB, et al. Potential roles for G-Quadruplexes in Mitochondria. Curr Med Chem. 2019;26:2918–2932.
- Falabella M, Kolesar JE, Wallace C, et al. G-quadruplex dynamics contribute to regulation of mitochondrial gene expression. Sci Rep. 2019;9:5605.
- Butler TJ, Estep KN, Sommers JA, et al. Mitochondrial genetic variation is enriched in G-quadruplex regions that stall DNA synthesis in vitro. Hum Mol Genet. 2020;29:1292–1309.
- Dahal S, Siddiqua H, Katapadi VK, et al. Characterization of G4 DNA formation in mitochondrial DNA and their potential role in mitochondrial genome instability. FEBS J. 2022;289:163–182.
- Li C-P, Huang J-H, Chang A-C, et al. G-quadruplex ligand 3,3’-diethyloxadicarbocyanine iodide induces mitochondrion-mediated apoptosis but not decrease of telomerase activity in nasopharyngeal carcinoma NPC-TW01 cells. Pharm Res. 2004;21:93–100.
- Huang W-C, Tseng T-Y, Chen Y-T, et al. Direct evidence of mitochondrial G-quadruplex DNA by using fluorescent anti-cancer agents. Nucleic Acids Res. 2015;43:10102–10113.
- Guo X, Yang D, Sun R, et al. A cyanine dye supramolecular FRET switch driven by G-quadruplex to monitor mitophagy. Dyes Pigm. 2021;192:109429.
- She M-T, Yang J-W, Zheng B-X, et al. Design mitochondria-specific fluorescent turn-on probes targeting G-quadruplexes for live cell imaging and mitophagy monitoring study. Chem Eng J. 2022;446:136947.
- Tseng T-Y, Wang Z-F, Chien C-H, et al. In-cell optical imaging of exogenous G-quadruplex DNA by fluorogenic ligands. Nucleic Acids Res. 2013;41:10605–10618.
- Tseng T-Y, Wang C-L, Huang W-C, et al. Folding and unfolding of exogenous G-Rich Oligonucleotides in live cells by fluorescence lifetime imaging microscopy of o-BMVC fluorescent probe. Molecules. 2021;27:140.
- Chang T, Qi C, Meng J, et al. General cell-binding activity of intramolecular G-quadruplexes with parallel structure. PLoS One. 2013;8:e62348.
- Yoon S, Rossi JJ. Aptamers: uptake mechanisms and intracellular applications. Adv Drug Deliv Rev. 2018;134:22–35.
- Sardiello M, Palmieri M. di Ronza A, Medina DL, Valenza M, Gennarino VA, Di Malta C, Donaudy F, Embrione V, Polishchuk RS, et al. Sci. 2009;325:473–477.
- Settembre C, Di Malta C, Polito VA, Garcia Arencibia M, Vetrini F, Erdin S, Erdin SU, Huynh T, Medina D, Colella P, et al. TFEB links autophagy to lysosomal biogenesis. Science. 2011;332:1429–1433.
- Beauvarlet J, Bensadoun P, Darbo E, et al. Modulation of the ATM/autophagy pathway by a G-quadruplex ligand tips the balance between senescence and apoptosis in cancer cells. Nucleic Acids Res. 2019;47:2739–2756.
- Miglietta G, Marinello J, Russo M, et al. Ligands stimulating antitumour immunity as the next G-quadruplex challenge. Mol Cancer. 2022;21:180.
- Moruno-Manchon JF, Lejault P, Wang Y, et al. Small-molecule G-quadruplex stabilizers reveal a novel pathway of autophagy regulation in neurons. Elife. 2020;9:e52283.
- Brozzi A, Urbanelli L, Germain PL, et al. hLGDB: a database of human lysosomal genes and their regulation. Database (Oxford). 2013;2013:bat024.
- Palmieri M, Impey S, Kang H et al. Characterization of the CLEAR network reveals an integrated control of cellular clearance pathways. Hum Mol Genet. 2011;20:3852–3866.
- Zhou W-J, Deng R, Zhang X-Y, et al. G-quadruplex ligand SYUIQ-5 induces autophagy by telomere damage and TRF2 delocalization in cancer cells. Mol Cancer Ther. 2009;8:3203–3213.
- Zhou W-J, Deng R, Feng G-K, et al. [A G-quadruplex ligand SYUIQ-5 induces autophagy by inhibiting the Akt-FOXO3a pathway in nasopharyngeal cancer cells]. Ai Zheng. 2009;28:1049–1053.
- Orlotti NI, Cimino-Reale G, Borghini E, et al. Autophagy acts as a safeguard mechanism against G-quadruplex ligand-mediated DNA damage. Autophagy. 2012;8:1185–1196.
- Beauvarlet J, Mergny J-L Djavaheri-Mergny, M. Activation of the Ataxia Telangiectasia Mutated/Autophagy pathway by a G-quadruplex ligand links senescence with apoptosis. Mol Cell Oncol. 2019;6:1604047.
- Ma X, Awadasseid A, Zhou K, et al. A 1,10-phenanthroline derivative selectively targeting telomeric G-quadruplex induces cytoprotective autophagy, causing apoptosis of gastric cancer cells. Life Sci. 2021;287:120095.
- Li L, Li Y, Zhao J, et al. CX-5461 induces autophagy and inhibits tumor growth via mammalian target of rapamycin-related signaling pathways in osteosarcoma. Onco Targets Ther. 2016;9:5985–5997.
- Bossaert M, Pipier A, Riou J-F, et al. Transcription-associated topoisomerase 2a (TOP2A) activity is a major effector of cytotoxicity induced by G-quadruplex ligands. eLife. 2021;10:e65184.
- Xu H, Hurley LH. A first-in-class clinical G-quadruplex-targeting drug. The bench-to-bedside translation of the fluoroquinolone QQ58 to CX-5461 (Pidnarulex). Bioorg Med Chem Lett. 2022;77:129016.
- Ismael M, Webb R, Ajaz M, et al. The Targeting of RNA polymerase I transcription using CX-5461 in combination with radiation enhances tumour cell killing effects in human solid cancers. Cancers (Basel). 2019;11:E1429.
- Chen H, Duo Y, Hu B, et al. PICT-1 triggers a pro-death autophagy through inhibiting rRNA transcription and AKT/mTOR/p70S6K signaling pathway. Oncotarget. 2016;7:78747–78763.
- Dannheisig DP, Schimansky A, Donow C, et al. Nucleolar stress functions upstream to stimulate expression of autophagy regulators. Cancers (Basel). 2021;13:6220.
- Sanchez-Martin V, Schneider DA, Ortiz-Gonzalez M, et al. Targeting ribosomal G-quadruplexes with naphthalene-diimides as RNA polymerase I inhibitors for colorectal cancer treatment. Cell Chem Biol. 2021;28(1590–1601.e4)
- Wang X, Zhang M, Xiong X-Q, et al. Design, synthesis and bioactivity of novel naphthalimide-benzotriazole conjugates against A549 cells via targeting BCL2 G-quadruplex and inducing autophagy. Life Sci. 2022;302:120651.
- Lejault P, Moruno-Manchon JF, Vemu SM, et al. Regulation of autophagy by DNA G-quadruplexes. Autophagy. 2020;16:2252–2259.
- Yousefi S, Perozzo R, Schmid I, et al. Calpain-mediated cleavage of Atg5 switches autophagy to apoptosis. Nat Cell Biol. 2006;8:1124–1132.
- Rubinstein AD, Eisenstein M, Ber Y, et al. The autophagy protein Atg12 associates with antiapoptotic Bcl-2 family members to promote mitochondrial apoptosis. Mol Cell. 2011;44:698–709.
- Onorati A, Dyczynski M, Ojha R, et al. Targeting autophagy in cancer. Cancer. 2018;124:3307–3318.
- Dumas L, Herviou P, Dassi E, et al. G-Quadruplexes in RNA biology: recent advances and future directions. Trends Biochem Sci. 2021;46:270–283.
- Burge S, Parkinson GN, Hazel P, et al. DNA: sequence, topology and structure. Nucleic Acids Res. 2006;34:5402–5415.