
ABSTRACT
Spautin-1 is a well-known macroautophagy/autophagy inhibitor via suppressing the deubiquitinases USP10 and USP13 and promoting the degradation of the PIK3C3/VPS34-BECN1 complex, while its effect on selective autophagy remains poorly understood. Mitophagy is a selective form of autophagy for removal of damaged and superfluous mitochondria via the autophagy-lysosome pathway. Here, we report a surprising discovery that, while spautin-1 remains as an effective autophagy inhibitor, it promotes PINK1-PRKN-dependent mitophagy induced by mitochondrial damage agents. Mechanistically, spautin-1 facilitates the stabilization and activation of the full-length PINK1 at the outer mitochondrial membrane (OMM) via binding to components of the TOMM complex (TOMM70 and TOMM20), leading to the disruption of the mitochondrial import of PINK1 and prevention of PARL-mediated PINK1 cleavage. Moreover, spautin-1 induces neuronal mitophagy in Caenorhabditis elegans (C. elegans) in a PINK-1-PDR-1-dependent manner. Functionally, spautin-1 is capable of improving associative learning capability in an Alzheimer disease (AD) C. elegans model. In summary, we report a novel function of spautin-1 in promoting mitophagy via the PINK1-PRKN pathway. As deficiency of mitophagy is closely implicated in the pathogenesis of neurodegenerative disorders, the pro-mitophagy function of spautin-1 might suggest its therapeutic potential in neurodegenerative disorders such as AD.
Disclaimer
As a service to authors and researchers we are providing this version of an accepted manuscript (AM). Copyediting, typesetting, and review of the resulting proofs will be undertaken on this manuscript before final publication of the Version of Record (VoR). During production and pre-press, errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal relate to these versions also.Introduction
Macroautophagy (referred as autophagy hereafter) is an evolutionarily conserved process for nonselective bulk degradation of proteins and organelles with important functions for maintenance of cellular homeostasis under stress conditions. The autophagic process consists of two consecutive stages: autophagosome biogenesis and autophagosome-lysosome fusion [Citation1–3]. These two stages are controlled by a group of proteins encoded by ATG (autophagy related) genes, the products of which form several complexes to execute the autophagic process [Citation4]. First, the ULK1 complex consisting of ULK1, ATG13, RB1CC1/FIP200 and ATG101 controls the initiation stage of autophagosome formation [Citation5,Citation6]. Second, the PIK3C3/VPS34 complex consisting of PIK3C3/VPS34-PIK3R4/VPS15, BECN1 (beclin 1), NRBF2 and ATG14 mediates the nucleation and expansion of the autophagosome precursor, the phagophore [Citation7]. Next, the two ubiquitination-like systems (ATG12 system and Atg8-family protein system) regulate the lipidation of MAP1LC3/LC3 (microtubule associated protein 1 light chain 3) [Citation8]. Finally, a protein complex inclusive of ATG14 and SNARE proteins (STX17 and SNAP29) controls the autophagosome-lysosome fusion [Citation9]. At present, numerous autophagic modulators have been developed by targeting different steps of the autophagic process for potential therapeutic application in human diseases [Citation10,Citation11]. Among them, spautin-1, a well-established autophagy inhibitor, is known to promote the degradation of BECN1 and PIK3C3/VPS34 by targeting the ubiquitin-specific peptidases, USP10 and USP13 [Citation12]. Suppression of these two deubiquitinating enzymes disrupts BECN1 and PIK3C3/VPS34 stability, consequently inhibiting autophagy [Citation12]. Inhibition of autophagy by spautin-1 promotes the degradation of mutant TP53/p53, leading to the death of cancer cells under nutritional deprivation conditions [Citation13]. In addition, spautin-1 shows anti-cancer functions, beyond suppression of autophagy, such as inhibiting mitochondrial complex I and NEK4 (NIMA related kinase 4) [Citation14–16].
Mitochondria are organelles with essential functions for maintaining normal cellular physiology including energy production, metabolism, thermogenesis, Ca2+ transport, redox signaling, inflammation and cell death [Citation17,Citation18]. Dysfunctional mitochondria are usually eliminated via various pathways to maintain the cellular homeostasis [Citation19–21]. Mitophagy is a selective form of autophagy for removal of damaged mitochondria via the autophagy-lysosome pathway. Among various mechanisms, the PINK1 (PTEN induced kinase 1)-PRKN (parkin RBR E3 ubiquitin protein ligase)-mediated mitophagy has been most extensively studied [Citation22,Citation23].
PINK1 has been well known as a sensor of mitochondrial damage to trigger mitophagy [Citation24]. Under physiological conditions, full length PINK1 (63 kDa) is transported into healthy mitochondria via the translocase of outer mitochondrial membrane (TOMM) complex consisting of TOMM70, TOMM20, TOMM22 and TOMM40, and TIMM23 (translocase of inner mitochondrial membrane 23) complex, and then cleaved by PARL (presenilin associated rhomboid like) at the inner mitochondrial membrane (IMM) [Citation25–28]. The resulting cleaved PINK1 (52 kDa) is released into the cytosol for degradation by the ubiquitin (Ub)-proteasome system (UPS) though the N-end rule [Citation29]. Under mitochondrial damaging conditions, the import of PINK1 is compromised without PARL-mediated cleavage, leading to its stabilization and accumulation at outer mitochondrial membrane (OMM) [Citation27,Citation30–32]. Stabilized PINK1 forms a homodimer and gains its kinase activity via autophosphorylation [Citation33]. The activated PINK1 then phosphorylates Ub at serine 65 (S65) and the ubiquitin-like(Ubl)domain of PRKN, leading a positive feedback loop via which activated PRKN is recruited from cytosol to damaged mitochondria where PRKN ubiquitinates a series of OMM proteins [Citation34–38]. Subsequently, specific mitophagy-adaptor proteins, such as OPTN (optineurin) and CALCOCO2/NDP52 (calcium binding and coiled-coil domain 2), are recruited to damaged mitochondria via binding to ubiquitinated OMM proteins, followed by binding to LC3-II at the phagophore membrane and eventually leading to the engulfment of damaged mitochondria within an autophagosome and degradation via a lysosome [Citation39–42]. It has been reported that these mitophagy receptors such as CALCOCO2 and OPTN are able to initiate autophagosome biogenesis via directly recruiting the autophagy machineries [Citation43,Citation44]. CALCOCO2 appears to recruit the ULK1/ULK2 complex via binding to RB1CC1/FIP200 to initiate the biogenesis of autophagosome [Citation44]. A more recent study reveals that distinct from CALCOCO2, OPTN utilizes the kinase TBK1 which directly binds to the BECN1-PIK3C3/VPS34 complex to initiate mitophagy even in the absence of ULK1 [Citation45].
Mitochondria play a fundamental role in health and life, including in neuronal function and resilience, while ageing and genetic mutations cause mitochondrial dysfunction and the accumulation of damaged mitochondria largely due to defective mitophagy [Citation46]. Thus, both mitochondrial dysfunction and compromised autophagy (especially mitophagy) are intertwined and are the two hallmarks of ageing, serving as causes/risks of common neurodegenerative diseases such as Alzheimer disease (AD), Parkinson disease (PD), amyotrophic lateral sclerosis [Citation46–50], and Huntington disease (HD) [Citation48–51]. There are multiple causes of defective mitophagy in these neurodegenerative diseases. For instance, mutations of gene encoding PINK1 or PRKN are associated with familiar early-onset PD [Citation52]. Also, accumulation of toxic proteins, including AD-related proteins Aβ and hyperphosphorylated MAPT/Tau (p-MAPT/tau), can interact with mitochondrial proteins to impair mitophagy [Citation53–55]. Moreover, aggregation of wild-type MAPT/Tau also translocate into mitochondria to disrupt mitophagy [Citation56,Citation57]. Therefore, one potential therapeutic strategy for treating such neurodegenerative disorders is to promote mitophagy [Citation47,Citation48,Citation58–61]. Herein, we unexpectedly found that, spautin-1, a well-known autophagy inhibitor, promotes PINK1-PRKN-mediated mitophagy. It also induces robust neuronal mitophagy in C. elegans and improves associative learning capability in an AD C. elegans model, highlighting its potential therapeutic applications for neurodegenerative disorders.
Results
Spautin-1 promotes PINK1-PRKN-mediated mitophagy upon mitochondrial damage.
Spautin-1 is a well-known autophagy inhibitor [Citation12]. In this study, we first confirmed the inhibitory effect of spautin-1 on the basal autophagy or autophagy induced by EBSS and torin-1, based on the reduction in MAP1LC3/LC3 puncta and the decrease in MAP1LC3/LC3 lipidation in SH-SY5Y cells expressing endogenous PRKN (Fig. S1A-1D). Interestingly, we observed that spautin-1 promoted LC3 lipidation induced by CCCP (Fig. S1E). We therefore investigated the effects of spautin-1 on mitophagy. To do this, we first checked the effect of spautin-1 on mitochondrial protein degradation, such as TOMM20, TIMM23 and COX4/COXIV (cytochrome c oxidase subunit 4) in HeLa cells stably expressing YFP-PRKN (YPH cells) and SH-SY5Y cells. The cells were treated with relatively low concentrations of carbonyl cyanide chlorophenylhydrazone (CCCP, 2.5 μM), oligomycin-antimycin (O/A, 125 nM/125 nM) and valinomycin (VAL, 2.5 nM) for long time (18 h), and we observed that treatment with spautin-1 markedly enhanced the reduction mitochondrial proteins induced by these mitochondrial damaging agents (; Fig. S1F), such effects were evident even when the cells were treated with lower concentration of spautin-1 (1 μM) (Fig. S1G). In comparison, treatment of these mitochondrial damaging agents (CCCP, O/A and VAL) alone at such low concentrations showed marginal changes of the mitochondrial proteins (Fig. S1H). In addition, we noticed that spautin-1 markedly enhanced mitochondrial damaging agents-induced cleavage of caspase 3, a hallmark of apoptosis (Fig. S1I). To investigate whether increased apoptotic signaling induced by spautin-1 had any effect on the reduction in IMM and OMM proteins, a pan-caspase inhibitor Z-VAD-FMK was used to prevent caspase-3 activation. We found that Z-VAD-FMK remarkably diminished cleaved caspase 3 without affecting the reduction of TIMM23, TOMM20 and COX4 induced by combination of mitochondrial damage agents with spautin-1 (Fig. S1I), indicating that reduction in IMM and OMM proteins by spautin-1 was not caused by caspases. It is well known that the mitophagy receptors such as CALCOCO2 are recruited to mitochondria upon induction of mitophagy [Citation40,Citation41]. Here we also observed that spautin-1 facilitated the translocation of CALCOCO2 to the mitochondria upon mitochondrial damage induced by CCCP (Fig. S1J-S1K). Together, these observations suggest that spautin-1 plays a positive role in regulation of mitophagy.
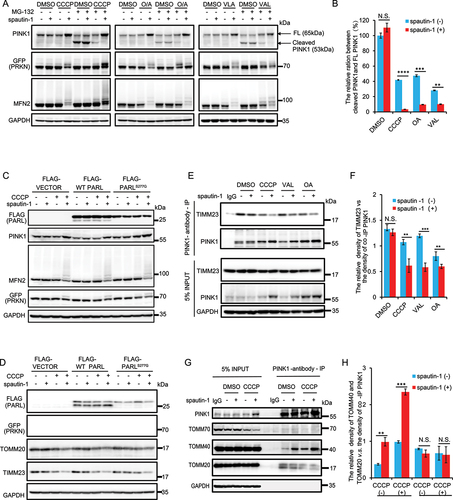
Figure 1. Spautin-1 promotes mitophagy. (A)YPH cells were pretreated with 20 μM or 40 μM spautin-1 for 6 h, and then treated with or without CCCP (2.5 μM), O/A (125 nM/125 nM) or valinomycin (VAL, 2.5 nM) for 18 h. The cell lysates were analyzed by western blotting using the indicated antibodies. (B) The quantification of mitochondrial OMM proteins (COX4, TIMM23 and TOMM20) in (A) were performed using ImageJ software. The data were presented as mean ± S.D. in three independent experiments (**, P < 0.01; ***, P < 0.001; ****, P < 0.0001, One-way ANVOA). (C) YPH-mt-Keima cells were pretreated with 40 μM spautin-1 for 6 h, and then treated with or without 5 μM CCCP for the indicated duration. The cells were then harvested for FACS analysis by excitation at 488 nm (neutral pH) and 561 nm (acidic pH). The gated area encloses cells undergoing mitophagy, and the percentage of cells within this gate is indicated in the top-left corner of each plot. (D) The percentage of cells undergoing mitophagy were quantified in cells treated as in (C). Data were presented as mean ± S.D. from three independent experiments (N.S., no significance; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001; Student’s t test). (E) YPH cells were pretreated with 20 μM spautin-1 for 6 h, and then treated with or without 2.5 μM CCCP for 18 h. The cells were then fixed with 4% PFA and subjected to immunofluorescence analysis after staining for anti-TOMM20 antibody and YFP-PRKN (green). The images were captured using a fluorescence microscope. Scale bar: 10 μm. The quantification data were presented in (F). (G) The relative intensity of mtDNA and DAPI (blue) were measured using immunofluorescence in the samples treated as in (E). The images were captured using a fluorescence microscope. The quantification data were presented in (H). For both F and H, data were presented as mean ± S.D. from 200 cells in three independent experiments (***, P < 0.001; ****, P < 0.0001, Student’s t test). (I) YPH cells were pretreated with 20 μM spautin-1 for 6 h, and then treated with or without 2.5 μM CCCP for 18 h. Relative mtDNA content was detected by real-time qPCR analysis of the mtDNA:nuclear DNA (nDNA) ratio (mtDNA:nDNA) in three independent experiments (***, P < 0.001, One-way ANVOA). (J) Oligomycin, FCCP, and antimycin A and rotenone were added sequentially into YPH cells treated with 20μM saputin-1 for 18 h. OCR and mitochondrial ATP production capacity was assessed by Seahorse Flux Analyzer, and the statistical data of ATP production were presented in (K). (L) Glucose, oligomycin and 2-DG were added sequentially into YPH cells treated with 20 μM spautin-1 for 18 h. ECAR was assessed by Seahorse Flux Analyzer.
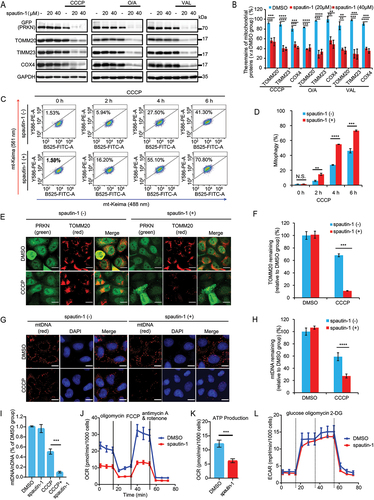
To further validate the positive effect of spautin-1 on mitophagy, we utilized the mitochondrial targeted Keima (mt-Keima) assay as a quantitative method to measure mitophagy based on the property of mt-Keima that it is a pH-sensitive protein and undergoes a gradual shift to longer-wavelength excitation within the acidic lysosome during mitophagy [Citation62]. We observed that there was a significant increase of mitophagy in cells with co-treatment of spautin-1 and CCCP in YPH-mt-Keima cells (). Measurement of TOMM20 or mitochondrial DNA (mtDNA) clearance is an alternative quantitative method for mitophagy assessment [Citation40,Citation63]. We then examined the level of TOMM20 and mtDNA and found that spautin-1 significantly reduced the level TOMM20 (, Fig. S1L-S1M) and mtDNA (, Fig. S1N-S1O) in cells treated with CCCP, O/A or VAL, respectively. Together, these data suggest that spautin-1 is capable of promoting mitophagy upon mitochondrial damage.
Finally, we assessed the effects of spautin-1 on mitochondrial functions including oxygen consumption rate (OCR), ATP production and extracellular acidification rate (ECAR) using Seahorse Flux Analyzer. Spautin-1 alone did not induce cell death nor had impact on cell proliferation (Fig. S1P-1Q). It reduces basal mitochondrial OCR and ATP production (), which is consistent with a previous report that spautin-1 inhibits the mitochondrial complex I [Citation16]. Additionally, spautin-1 had no influence on ECAR, suggesting spautin-1 does not impact glycolysis ().
Spautin-1 promotes mitochondrial recruitment and activation of PRKN upon mitochondrial depolarization.
The recruitment of PRKN to damaged mitochondria and its activation is an essential step in mitophagy [Citation35,Citation38]{Kondapalli, 2012 #36}{Kondapalli, 2012 #36}. To examine the mechanisms underlying the pro-mitophagy activity of spautin-1, here we asked if spautin-1 affects PRKN recruitment upon mitochondrial damage. We first examined the mitochondrial translocation of PRKN upon CCCP treatment and found that PRKN was recruited to the mitochondria as early as 0.5 h in the presence of spautin-1, while treatment of CCCP alone induced PRKN mitochondrial translocation to a lesser extent (). Similar results were observed when mitochondria were damaged by O/A or VAL (Fig. S2A-S2B). Next, we evaluated whether PRKN E3 ligase activity was affected by spautin-1 upon mitochondrial damage. As OMM proteins undergo ubiquitination mediated by PRKN upon mitophagy induction [Citation41], we examined the ubiquitination levels of OMM proteins in the presence or absence of spautin-1. As expected, addition of spautin-1 markedly elevated the ubiquitination level of OMM proteins including MFN1 and MFN2 (, Fig. S2C). Similar results were found in SH-SY5Y cells (Fig. S2D). Such effects were even observed at different concentrations of spautin-1(Fig. S2E). To further solidify our findings, we pulled down MFN2 and confirmed the enhanced ubiquitination of MFN2 by spautin-1 in cells treated with CCCP ().
Figure 2. Spautin-1 promotes mitochondrial translocation of PRKN and activates its E3 ligase activity. (A) YPH cells were pretreated with 20 μM spautin-1 for 6 h, and then treated with or without 5 μM CCCP for indicated duration. The cells were subjected to immunofluorescence analysis for PRKN (green) and MitoTracker. Scale bar: 10 μm. The images were captured using the confocal microscope. The number of PRKN mitochondrial translocation per cell was quantified by counting 200 cells from three independent experiments and presented mean ± S.D. as in (B) (***, P < 0.001; ****, P < 0.0001, One-way ANVOA). (C) YPH cells were treated as in (A) and subjected to western blotting analysis. (D) YPH cells were pretreated with 20 μM or 40 μM spautin-1 for 6 h, and then treated with or without 5 μM CCCP for indicated time points. Cells were lysed with IP lysis buffer and subjected to IP using MFN2 antibody and analyzed with the indicated antibodies. (E) The relative density of polyubiquitination of MFN2 in (D) was analyzed using ImageJ software and the data were presented as mean ± S.D. in three independent experiments (**, P < 0.01; ***, P < 0.001; ****, P < 0.0001, One-way ANVOA). (F) YPH cells were treated as in (D). Cells were subjected to IP using GFP agarose and analyzed with the indicated antibodies. (G) The quantification of relative density of polyubiquitination of PRKN in (F) was performed using ImageJ software. The data were presented as mean ± S.D. in three independent experiments (**, P < 0.01; ***, P < 0.001; ****, P < 0.0001, One-way ANVOA).
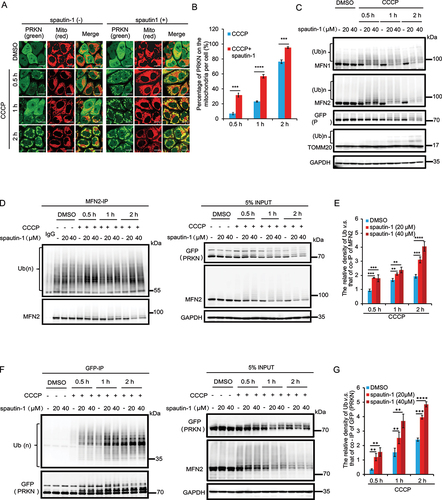
It is known that activation of PRKN promotes auto-ubiquitination and degradation of PRKN [Citation64]. We thus evaluated the ubiquitination and protein level of PRKN and observed a remarkable increase in ubiquitination and a significant reduction in the protein level of PRKN following treatment of spautin-1 plus CCCP (). Collectively, these data support the notion that spautin-1 accelerates the mitochondrial recruitment of PRKN and enhances its E3 ligase activity upon mitochondrial damage.
Spautin-1 promotes PINK1-PRKN-mediated mitophagy independent of USP10 and USP13.
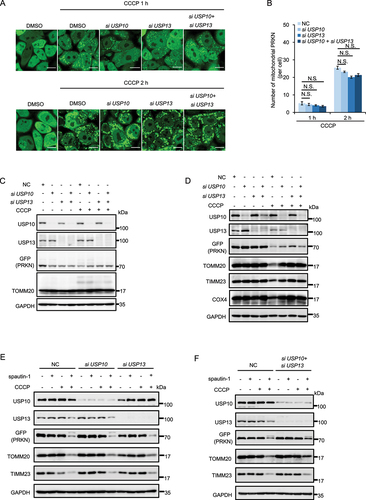
It has been reported that spautin-1 suppresses autophagy via inhibition of USP10 and USP13, the two deubiquitinases to degrade PIK3C3/VPS34 and BECN1 [Citation12]. We wondered whether the effects of spautin-1 on mitophagy were related to its known inhibitory effect on USP10 and USP13. In this part of our study, we found that knockdown of USP10 or/and USP13 did not enhance the recruitment of PRKN to mitochondria nor increase its E3 ligase activity (). As expected, knockdown of USP10 or/and USP13 did not promote CCCP-induced mitophagy (). More importantly, the absence of USP10 or/and USP13 did not change the promoting effect of spautin-1 on CCCP-induced mitophagy (). On the other hand, various deubiquitinases (such as USP8, USP15 and USP30) have been reported to negatively regulate mitophagy via antagonizing PRKN-mediated ubiquitination [Citation65–67]. We thus asked whether these deubiquitinases are implicated in the mitophagy promoting activity of spautin-1. As shown in Fig. S3, knockdown of USP8 and USP15 failed to affect the effects of spautin-1. Therefore, it is believed that the regulatory role of spautin-1 in mitophagy is independent of these deubiquitinases.
Figure 3. Spautin-1 promotes PINK1-PRKN-mediated mitophagy independent on USP10 and USP13. (A) YPH cells were transfected with siUSP10, siUSP13 and siUSP10 plus siUSP13 for 48 h. The cells were then treated with 5 μM CCCP for indicated time point and subjected to the confocal microscope analysis. Percentage of cells with PRKN mitochondrial translocation was quantified by counting 200 cells from three independent experiments and presented mean ± S.D. as in (B) (N.S., No significance, Student’s t test). Scale bar: 10 μm. (C) YPH cells were transfected with siUSP10, siUSP13 and siUSP10 plus siUSP13 for 48 h. The cells were treated with 5μM CCCP for 2 h. The cells were harvested and subjected to Western blotting analysis. (D) YPH cells were transfected with siUSP10, siUSP13 and siUSP10 plus siUSP13 for 48 h. The cells were then treated with 2.5 μM CCCP for 18 h and subjected to western blotting analysis. (E and F) YPH cells were transfected with siUSP10, siUSP13 and siUSP10 plus siUSP13 for 48 h. The cells were pretreated with 20 μM spautin-1 for 6 h, and then treated with or without 2.5 μM CCCP for 18 h. The cells were harvested and subjected to western blotting analysis.
Spautin-1 facilitates PINK1 stabilization and activation in damaged mitochondria.
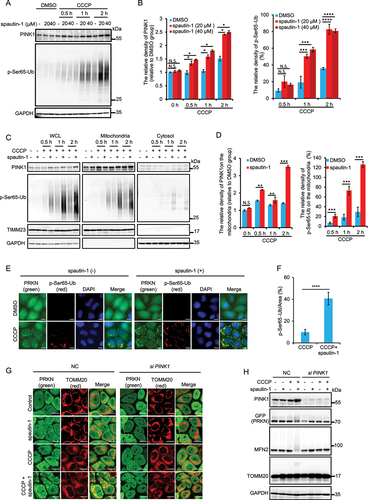
Based on the well-known fact that activation of PRKN is dependent on PINK1 via PINK1-mediated phosphorylation of both PRKN and Ub [Citation35,Citation36,Citation38,Citation68–70], we therefore examined the effects of spautin-1 on PINK1 stability upon mitochondrial damage. While mitochondrial damaging agents (CCCP, O/A and VAL) effectively stabilized full length PINK1, this effect was enhanced by spautin-1 (, Fig. S4A-S4C). Consistently, PINK1 mediated phosphorylation of Ub at Ser65 (p-Ser65-Ub) was also remarkably increased (, Fig. S4A). Moreover, we observed significant accumulation of full-length PINK1 and p-Ser65-Ub in the mitochondrial fraction isolated from YPH cells with spautin-1 plus CCCP treatment (). These findings were further confirmed by immunofluorescent staining or Western blotting that showed marked increase of p-Ser65-Ub in YPH cells (, Fig. S4D-S4E) or in SH-SY5Y cells (Fig. S4F-S4G). Upon PINK1 knockdown, spautin-1 failed to promote PRKN mitochondrial translocation () and subsequent PRKN E3 ligase activation evidenced by suppression of poly-ubiquitination of MFN2 (). Thus, data from this part of our study suggest that the effects of spautin-1 on PRKN mitochondrial translocation and activation are PINK1-dependent and that spautin-1 facilitates the stabilization and activation of PINK1 upon mitochondrial damage.
Figure 4. Spautin-1 stabilizes and activates PINK1. (A) YPH cells were pretreated with 20 μM or 40 μM spautin-1 for 6 h, and then treated with or without 5 μM CCCP for indicated time points. The cells were harvested and subjected to western blotting analysis. The relative density of PINK1 and p-Ser65-Ub were presented. The relative density of PINK1 and p-Ser65-Ub in (A) were analyzed using ImageJ software, and the data were presented as mean ± S.D. in three independent experiments (**, P < 0.01; ***, P < 0.001; ****, P < 0.0001; N.S., no significance, One-way ANVOA) in (B). (C) Cell fraction assay was performed to isolate mitochondria from YPH cells treated with 5 μM CCCP and 20 μM spautin-1 for indicated time points. The whole cell lysate (WCL), mitochondrial fraction and cytosol were then subjected to western blotting analysis with indicated antibodies. (D) The relative density of PINK1 and p-Ser65-Ub on the mitochondrial fraction were analyzed using ImageJ software, and the data were presented as mean ± S.D. in three independent experiments (**, P < 0.01; ***, P < 0.001; N.S., no significance, Student’s t test). (E) The cells were pretreated with 20 μM spautin-1 for 6 h, and then treated with or without 5 μM CCCP for 1 h. The cells were then fixed with 4% PFA and subjected to immunofluorescence analysis after staining for p-Ser65-Ub antibody and DAPI (blue). The images were captured using the fluorescence microscope. Scale bar: 10 μm. (F) The relative intensity of p-Ser65-Ub in the samples treated as in (E) was presented as mean ± S.D. from 200 cells in three independent experiments (****, P < 0.0001, Student’s t test). (G) YPH cells were transfected with siNC and siPINK1 for 48 h. The cells were pretreated with 20 μM spautin-1 for 6 h, and then co-treated with or without 5 μM CCCP for 1 h. The cells were then fixed with 4% PFA and subjected to the confocal microscope analysis for TOMM20 and PRKN (green). Scale bar: 10 μm. (H) Cells were treated as in G and then subjected to Western blotting using indicated antibodies.
Spautin-1 prevents the import and cleavage of PINK1.
Having established that spautin-1 promotes mitophagy via stabilization of PINK1, we next aimed to study the underlying mechanisms. The stabilization of PINK1 at OMM is induced by mitochondrial depolarization [Citation36]. We thus evaluated the effects of spautin-1 on mitochondrial membrane potential (MMP) using the TMRE dye which accumulates only in healthy mitochondria but not in the depolarized mitochondria. We first found that spautin-1 did not affect MMP with or without CCCP treatment (Fig. S5A-S5C), indicating that the stabilization of PINK1 by spautin-1 is not due to MMP alteration. In normal and unstressed mitochondria, PINK1 is imported into mitochondria via the TOMM complex, cleaved by PARL at IMM and the cleaved PINK1 is extracted to the cytosol for proteasomal degradation to maintain low PINK1 protein level [Citation25,Citation28,Citation31]. Therefore, we attempted to explore whether spautin-1 is capable of influencing the import and cleavage of PINK1 to promote its stabilization at the OMM. As shown in , addition of MG-132, a proteasome inhibitor, markedly increased cleaved PINK1 in control cells. Importantly, spautin-1 not only enhanced the full-length PINK1 protein level in YPH cells treated with CCCP, O/A or VAL, it also reduced cleaved PINK1 in those cells treated with MG-132. In addition, treatment with spautin-1 alone had marginal effects on cleaved PINK1 in the control cells (). Similar results were also found in SH-SY5Y cells (Fig. S5D). All these results substantiate the notion that spautin-1 may impede the cleavage of PINK1 to facilitate the stabilization of full length PINK1 at damaged mitochondria.
Figure 5. Spautin-1 prevents the import and cleavage of PINK1. (A) YPH cells were pretreated with 20 μM spautin-1 for 6 h, and then treated with or without CCCP (5 μM), O/A (250 nM/250 nM) and VAL (5 nM), together with MG-132 (10 μM) for 2 h. The cells were harvested and subjected to western blotting analysis. (B) The ratio between cleaved PINK1 and full-length PINK1 in the samples treated as in (A) was analyzed using ImageJ software. The data were presented as mean ± S.D. in three independent experiments (**, P < 0.01; ***, P < 0.001; ****, P < 0.0001; N.S., no significance, Student’s t test). (C) YPH cells were transfected with FLAG-PARL wildtype (WT) or FLAG-PARL catalytic mutant (S277G) for 24 h. The cells were pretreated with 20 μM spautin-1 for 6 h, and then treated with or without 5 μM CCCP for 1 h. Lysate were subjected to western blotting analysis. (D) YPH cells were transfected with FLAG-PARL wildtype (WT) or FLAG-PARL catalytic mutant (S277G) for 24 h. The cells were pretreated with 20 μM spautin-1 for 6 h, and then treated with or without 2.5 μM CCCP for 18 h. Lysate were subjected to western blotting analysis. (E) YPH cells were pretreated with 20 μM spautin-1 for 6 h, and then co-treated with or without CCCP (5 μM), VAL (5 nM) and O/A (250 nM/250 nM) for 2 h. Cells were lysed with IP lysis buffer and subjected to IP using PINK1 antibody and analyzed with the indicated antibodies. (F) The relative density of TIMM23 relative to co-IP of PINK1 in (E) was analyzed using ImageJ software, and the data were presented as mean ± S.D. in three independent experiments (**, P < 0.01; ***, P < 0.001; N.S., no significance, Student’s t test). (G) YPH cells were treated as in (F), and then lysed with IP lysis buffer. After that, the lysates were subjected to IP using PINK1 antibody and analyzed with the indicated antibodies. (H) The quantification of the relative density of TOMM20 and TOMM40 relative to co-IP of PINK1 in (G) were analyzed using ImageJ software, and the data were presented as mean ± S.D. in three independent experiments (**, P < 0.01; ***, P < 0.001; N.S., no significance, Student’s t test).
Blockage of PINK1 import into IMM and inactivation of hydrolase PARL are the two critical steps contributing to PINK1 stability [Citation28]. To further assess the effect of spautin-1 on PINK1 cleavage and stabilization, we overexpressed both wild-type (WT) PARL and catalytic mutant (S277G) PARL, and found that spautin-1 had similar effects on PINK1 stabilization and PRKN activation regardless of PARL status (). Consistently, the effect of spautin-1 on mitophagy was also independent of PARL activity, as evidenced by the comparable reduction of both TOMM20 and TIMM23 protein level in cells overexpressing mutated PARL (). Together, these data suggest that the effect of spautin-1 on PINK1 is not related to PARL activity. Next, we asked whether spautin-1 impairs the import of PINK1. To address this point, we examined the interaction between PINK1 and TIMM23, a critical component of the TIM complex responsible for the import of PINK1 [Citation30]. As expected, treatment with mitochondrial damaging agents (CCCP, O/A or VAL) impeded the interaction between PINK1 and TIMM23, and spautin-1 further weakened their interaction (). Upon induction of mitophagy, PINK1 is stabilized by anchoring to the OMM with the help of TOMM complex [Citation27,Citation30–32]. Here, we further examined the interaction between PINK1 and main components of TOMM complex including TOMM20, TOMM40 and TOMM70. Among them, TOMM40 is a channel-forming protein and form a pore into the mitochondria, which is critical for stable interaction of PINK1 with the TOMM complex [Citation27]. We observed that spautin-1 strengthened the interaction of PINK1 with TOMM40 in CCCP-treated cells (), while binding of PINK1 with TOMM20 or TOMM70 is weaker, consistent with a previous study [Citation27,Citation32]. Unexpectedly, even though spautin-1 alone also increased the interaction between TOMM40 and PINK1 (), the level of full-length PINK1 or cleaved PINK1 was not influenced under basal conditions (). Taken together, these data demonstrate that spautin-1 retards the import of PINK1 to facilitate its stability upon mitochondrial damage.
Spautin-1 biochemically interacts with the TOMM complex.
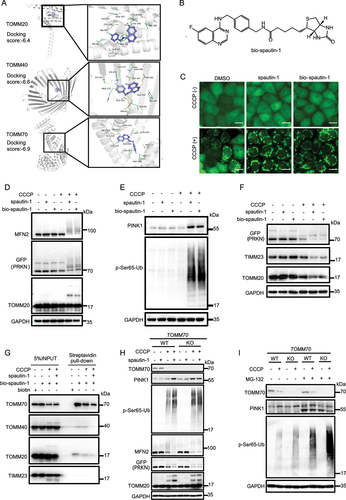
To further understand how spautin-1 blocks the import of PINK1, we examined the target proteins of spautin-1 by molecular docking, a tool to assess the binding affinity between drugs and ligand molecules [Citation71]. We found that spautin-1 had higher scores with selected TOMM complex proteins (), suggesting that spautin-1 may target the TOMM complex to modulate the PINK1 stability. It is known that TOMM20, TOMM40 and TOMM70, the 3 key proteins in the TOMM complex, play vital roles in the import and stabilization of PINK1 [Citation30,Citation72–74]. Through generating the molecular docking model using AUTODOCK VINA 1.1.2 software [Citation71], we obtained the possible binding sites of spautin-1 on TOMM20, TOMM40 and TOMM70 as shown in . We noticed that these binding sites covered the functional regions of these three core proteins [Citation75,Citation76]. To verify this prediction, we first synthesized spautin-1 conjugated with biotin (thereafter named as bio-spautin-1) (), and found that bio-spautin-1 had similar activity as spautin-1 to promote mitochondrial translocation of PRKN, stabilization of PINK1 and activation of mitophagy (, Fig. S6A-S6B) [Citation27]. Next, using streptavidin-biotin affinity-isolation assay, we found that bio-spautin-1 interacted with the main components of TOMM complex, including TOMM20, TOMM40 and TOMM70, while its binding to TIMM23 was undetectable (). Furthermore, unlabeled spautin-1 could competitively bind to these proteins (), further supporting that the TOMM complex were the target proteins of spautin-1. Of note, bio-spautin-1 had a much stronger affinity with TOMM70 than with TOMM20 or TOMM40 (). Although the interaction between TOMM70 with PINK1 is marginal (), TOMM70 has been widely reported as an essential component of the TOMM complex for PINK1 import to mitochondria [Citation27,Citation72,Citation75,Citation77]. Consistently, we established the TOMM70 KO cells and found that TOMM70 KO promoted PINK1 accumulation and subsequent mitochondrial translocation of PRKN and its E3 ligase activity (, Fig. S6C-S6D). Of note, we did not find additive effects of spautin-1 on PINK1 stabilization in TOMM70 KO cells (). Additionally, we observed a slight reduction of cleaved PINK1 in TOMM70 KO cells under the mitochondrial stress condition (), similar to the effect of spautin-1 (). Such observations clearly indicate the importance of TOMM70 in the import of PINK1. Together, these data support the notion that spautin-1 possibly facilitate the stabilization of PINK1 via targeting the core proteins of the TOMM complex upon mitochondrial depolarization.
Figure 6. Spautin-1 directly binds to the TOMM complex. (A) Structural illustration showing possible binding of spautin-1 with TOMM complex (TOMM20, TOMM40 and TOMM70). (B) The structure of bio-spautin-1 was presented. (C) YPH cells were pretreated with bio-spautin-1 (20 μM) or spautin-1 (20 μM) for 6 h, and then treated with or without 5 μM CCCP for 1 h. The cells of PRKN with mitochondrial translocation were observed under the fluorescence microscope. Scale bar: 20 μm. (D and E) YPH cells were pretreated with 20 μM bio-spautin-1 or spautin-1 for 6 h, and then treated with or without 5 μM CCCP for 1 h. The cells were harvested and subjected to western blotting analysis. (F) YPH cells were pretreated with 20 μM bio-spautin-1 or spautin-1 for 6 h, and then treated with or without 2.5 μM CCCP for 18 h. The cells were harvested and subjected to western blotting. (G) YPH cells were treated with or without 5 μM CCCP for 1 h. The cells were harvested and lysed with IP lysis buffer. Then, lysate was subjected to streptavidin-biotin affinity-isolation using streptavidin magnetic beads and analyzed with the indicated antibodies. (H) YPH cells and YPH TOMM70 KO cells were pretreated with 20 μM spautin-1 for 6 h, and then treated with or without 5 μM CCCP for 1 h. The cells were harvested and subjected to western blotting analysis with indicated antibodies. (I) YPH cells and YPH TOMM70 KO cells were were pretreated with 20 μM spautin-1 for 6 h, and then treated with or without 5 μM CCCP together with MG-132 (10 μM) for 1 h. The cells were harvested and subjected to western blotting analysis.
Spautin-1 is capable of improving the neuronal function of HsMAPT/Tau-AD C. elegans by inducing mitophagy.
After establishing the pro-mitophagy function of spautin-1 in vitro, we used the AD-like HsMAPT/TauP301L C. elegans as animal model to evaluate whether spautin-1 can promote mitophagy in vivo. These AD worms recapitulate some key features of AD patients, including neurodegeneration, associative learning capability loss, and shorter lifespan [Citation48,Citation61]. Based on the toxicity assay, we first identified 1 μM of spautin-1 as a safe dose for the nematodes in the subsequent assays (Fig. S7A-S7D). We tested the effect of spautin-1 on mitophagy using transgenic nematodes with pan-neuronal mitochondria-targeted Rosella (mt-Rosella, a dual-color-emission biosensor). The mt-Rosella biosensor consists two proteins including a lysosomal lumen-sensitive green fluorescent protein (GFP) variant and a pH-insensitive DsRed [Citation48,Citation61,Citation62]. GFP:DsRed ratio is used to assess mitophagy index, with reduced values representing mitophagy induction (). We verified the presence of mitophagy impairment in HsMAPT/TauP301L;n-sid-1OV;mt-Rosella C. elegans (). Subsequently, our results demonstrated that spautin-1 was able to promote neuronal mitophagy in N2n-sid-1OV;mt-Rosella worms ( and Fig. S7E). To understand the molecular mechanisms responsible for the pro-mitophagy by spautin-1, we knocked down neuronal pink-1 (the C. elegans homolog of the mammalian PINK1) and pdr-1 (the C. elegans homolog of the mammalian PRKN) via the well-established RNAi feeding approach. Depletion of pink-1 and pdr-1 in the neuron system eliminated spautin-1 induced mitophagy in N2n-sid-1OV;mt-Rosella ( and Fig. S7E). Collectively, spautin-1 promotes neuronal mitophagy via a PINK1- and PRKN-dependent manner in C. elegans.
Figure 7. Spautin-1 promotes mitophagy and restores associative learning capability deficits in the AD-like HsMAPT/TauP301L C. elegans model. (A) Schematic working model of the mt-Rosella expressed in the pan-neuronal system of the C. elegans (Ex[punc-119TOMM-20:Rosella;rol-6(su1006)]). Mitophagy index is calculated as GFP:DsRed ratio, thus lower values correlate with higher mitophagy. (B) A worm with enhanced neuronal mitophagy is shown (the boxed area highlights tail neurons). To eliminate confounding signals (worm gut autofluorescence and the signal of co-injection marker from pharynx), we quantified mitophagy signal in neurons of the tail region. (C-E) Effects of 1 μM spautin-1 on the induction of neuronal mitophagy in worms with mt-Rosella reporter at adult day 2. Data were pooled from three biological replicates (total n = 30 ~ 53 nematodes per group). Kruskal-Wallis test followed by Multiple comparisons was used for data analysis. (F) Effects of 1 μM spautin-1 on associative learning capability in transgenic animals expressing HsMAPT/TauP301L;n-sid-1OV at adult day 2 and the effects of pink-1, pdr-1, lgg-1 on spautin-1-dependent associative learning capability improvement in the HsMAPT/TauP301L;n-sid-1OV worms at adult day 2. Two-way ANOVA followed by Tukey’s multiple comparisons test was used for data analysis. (G) Effect of 1 μM spautin-1 on lifespan in both WT and HsMAPT/TauP301L worms. Survival analysis followed by Log-rank (Mantel-Cox) was used for data analysis. All quantitative data are shown in mean ± S.D. from two to three biological repeats (N.S., no significance; *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001).
![Figure 7. Spautin-1 promotes mitophagy and restores associative learning capability deficits in the AD-like HsMAPT/TauP301L C. elegans model. (A) Schematic working model of the mt-Rosella expressed in the pan-neuronal system of the C. elegans (Ex[punc-119TOMM-20:Rosella;rol-6(su1006)]). Mitophagy index is calculated as GFP:DsRed ratio, thus lower values correlate with higher mitophagy. (B) A worm with enhanced neuronal mitophagy is shown (the boxed area highlights tail neurons). To eliminate confounding signals (worm gut autofluorescence and the signal of co-injection marker from pharynx), we quantified mitophagy signal in neurons of the tail region. (C-E) Effects of 1 μM spautin-1 on the induction of neuronal mitophagy in worms with mt-Rosella reporter at adult day 2. Data were pooled from three biological replicates (total n = 30 ~ 53 nematodes per group). Kruskal-Wallis test followed by Multiple comparisons was used for data analysis. (F) Effects of 1 μM spautin-1 on associative learning capability in transgenic animals expressing HsMAPT/TauP301L;n-sid-1OV at adult day 2 and the effects of pink-1, pdr-1, lgg-1 on spautin-1-dependent associative learning capability improvement in the HsMAPT/TauP301L;n-sid-1OV worms at adult day 2. Two-way ANOVA followed by Tukey’s multiple comparisons test was used for data analysis. (G) Effect of 1 μM spautin-1 on lifespan in both WT and HsMAPT/TauP301L worms. Survival analysis followed by Log-rank (Mantel-Cox) was used for data analysis. All quantitative data are shown in mean ± S.D. from two to three biological repeats (N.S., no significance; *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001).](/cms/asset/2e6cd3bb-63fe-4ce2-bc2a-a4e16ca1e25b/kaup_a_2383145_f0007_c.jpg)
We next tested whether spautin-1 has associative learning capability restoration capacity in C. elegans, based on the well-known fact that compromised mitophagy pathway is implicated in AD progression and pathogenesis [Citation48,Citation49,Citation61,Citation78]. We first confirmed that mitophagy in the AD-like HsMAPT/TauP301L;n-sid-1OV;mt-Rosella worms is indeed compromised (). We then found that spautin-1 was able to upregulate mitophagy in the HsMAPT/TauP301L;n-sid-1OV;mt-Rosella worms, and such effect was abolished upon depletion of pink-1 or prd-1 ( and Fig. S7E). To study the effect of spautin-1 on the associative learning capability preservation capacity of AD worm models, a chemotaxis assay was performed for the simple associative learning capability evaluation in nematodes, with lower score correlates with better associative learning capability. Accordingly, animals treated with spautin-1 demonstrated improved associative learning capability in HsMAPT/TauP301L;n-sid-1OV worms (). Consistently, such spautin-1-induced associative learning capability improvement was fully dependent on pink-1 and pdr-1 (). Given the pivotal role of MAP1LC3/LC3 in both autophagy and mitophagy, we also knocked down lgg-1 (the ortholog of the mammalian protein LC3). Such effect of spautin-1 was abolished upon depletion of lgg-1 ().
We further tested the effect of spautin-1 on the lifespan in the nematode. The HsMAPT/TauP301L worms exhibited a reduced lifespan in comparison to the WT N2 worms (). However, the administration of spautin-1 extended the average lifespan of WT N2 nematodes by 14.29% and increased the lifespan of HsMAPT/TauP301L nematodes by 11.11% (). Data from this part of our study demonstrate that spautin-1 forestalls associative learning capability loss in the HsMAPT/Tau AD worms via stimulating pink-1-pdr-1-dependent mitophagy.
Discussion
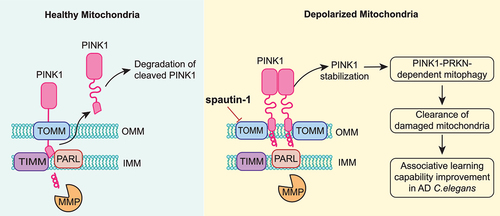
In this study, we discovered a novel function of spautin-1 in mitophagy. spautin-1 is capable of promoting PINK1-PRKN-mediated mitophagy induced by mitochondrial damaging agents. Mechanistically, spautin-1 promotes stabilization of full-length PINK1 at OMM via directly binding to the core proteins of TOMM complex, leading to impairment of PINK1 import and cleavage of PINK1 by PARL. Functionally, spautin-1 is capable of enhancing neuronal mitophagy and improved associative learning capability in AD nematodes model. In summary, we reveal an unexpected finding: while known as an effective inhibitor of autophagy, spautin-1 is capable of promoting mitophagy via PINK1 stabilization and improve neuronal function ().
Figure 8. Schematic model for the positive regulatory role of spautin-1 in mitophagy.
Spautin-1 is well-known to inhibit autophagy via suppressing the BECN1- PIK3C3/VPS34 complex by directly inhibiting the activity of USP10 and USP13, two deubiquitinase that stabilize PIK3C3/VPS34 and BECN1 [Citation12]. In this study, we observed an unexpected effect of spautin-1 on promoting mitophagy in cells treated with mitochondrial damaging agents (CCCP, O/A and VAL), based on the following results: First, spautin-1 accelerates the degradation and clearance of mitochondrial proteins (; Fig. S1). Second, spautin-1 stimulates PRKN translocation and activity (; Fig. S2). Third, spautin-1 facilitates the stabilization and activation of PINK1 at OMM of damaged mitochondria (; Fig. S4). One intriguing question here is how the pro-mitophagy effects of spautin-1 overrides its inhibition on BECN1- PIK3C3/VPS34-dependent autophagy. In this study, we confirmed that spautin-1 could strikingly impaired MAP1LC3/LC3 lipidation even under basal condition, amino acid starvation (EBSS) or MTORC1 inhibition (torin-1) conditions (Fig. S1A-S1D). In contrast, spautin-1 further enhanced MAP1LC3/LC3 lipidation in cells treated with CCCP (Fig. S1E). Given that BECN1-independent autophagy could be induced under various conditions [Citation79,Citation80], one possible explanation for such intriguing findings is that spautin-1 may engage other autophagy machinery independent of BECN1-PIK3C3/VPS34 in the process of mitophagy. More work is required to study the detailed mechanisms.
Activation of PINK1 is a critical event in the course of mitophagy. Here we showed that spautin-1 accelerated the stabilization of PINK1 upon mitochondrial damage ( and Fig. S4). It has been reported that the stabilization of PINK1 is caused by the loss of MMP which blocks PINK1 mitochondrial import, prevents its cleavage by PARL, and anchors PINK1 in the TOMM complex, leading to accumulation and retention of PINK1 at OMM [Citation26,Citation30,Citation31]. Here, we showed that spautin-1 has marginal effect on MMP (Fig. S5A-S5C). In normal and unstressed mitochondria, association of PINK1 with TOMM complex allows the import of PINK1 into IMM and mitochondrial matrix, a step required for cleavage of PINK1 by PARL [Citation31]. Our results showed that spautin-1 influences the import of PINK1, instead of its cleavage, to strengthen its stability at the OMM ( -). It is worth noting that the effect of spautin-1 on PINK1 accumulation is only evident in cells treated with mitochondrial damaging agents, but not under basal conditions (), which is consistent with the fact that spautin-1 alone has no evident effect on MMP (Fig. S5A-S5C). Thus, it is believed that the effect of spautin-1 on PINK1 import in terms of magnitude is much weaker than that of mitochondrial damaging agents such as CCCP, and thus it is unable to possess any significant effect on PINK1 under basal conditions. Similarly, spautin-1 may have minimal effect on the import of other mitochondrial proteins, although the details are subject to further investigation.
TOMM70 has been described as a main import receptor for PINIK1 and deletion of TOMM70 increases mitophagy flux [Citation43]. In this study, spautin-1 is able to bind to various subunits of the TOMM complex including TOMM20, TOMM40 and TOMM70 (). Among these subunits, spautin-1 has much stronger affinity to TOMM70 (). The depletion of TOMM70 could produce similar effects on PINK1 stability as spautin-1 (), and the addictive effects of spautin-1 on the PINK1 stability and PRKN E3 ligase activity upon mitochondrial damage was abolished in TOMM70 KO cells (). These findings thus suggest that TOMM70 is a major target for spautin-1 to promote mitophagy. We further predicted that spautin-1 can form hydrogen bound with LEU207, MET225, ALA228, LEU232, ALA590, LEU587, PHE568 and CAL556 (), and these binding sites cover the docking region for the hydrophobic substrates of TOMM70 (aa 235-608) [Citation75,Citation81]. These findings thus reveal a novel function of spautin-1 in regulation of PINK1 stability via interaction with the TOMM complex. Further investigation to specify the functional regions of TOMM20, TOMM40 and TOMM70 for spautin-1 binding would be important for the optimization of the spautin-1 structure to target mitophagy more specifically.
Defective mitophagy and the resulted accumulation of dysfunctional mitochondria is a possible underlying mechanism for neurodegenerative diseases such as AD and PD [Citation49,Citation60]. While so far there are no effective treatments for such diseases, various therapeutic interventions targeting mitophagy have been explored to delay the disease progression [Citation48,Citation59]. Mitophagy inducers have been reported to promote the survival of neurons, alleviate amyloid-β and MAPT/tau pathologies, and improve the memory in AD animal models [Citation48,Citation82]. In this study, we found that spautin-1 is able to promote neuronal mitophagy and improve associative learning capability in AD nematodes (). Such effects were dependent on PINK1 and PRKN, consistent with our findings in the mammalian cell model in vitro. This is also consistent with some previous studies in which pharmacological or genetic PINK1 activation promotes the clearance of damaged mitochondria by augmenting mitophagy to ameliorate pathology in PD model or AD model [Citation47,Citation59]. Our findings thus support the neuroprotective role of the PINK1-PRKN pathway in AD by illustrating spautin-1‘s regulation of PINK1-PRKN-dependent mitophagy that leads to the restoration of associative learning capabilities in vivo (). Our data are in line with the known effects of PINK1 and PRKN on synaptic health, structure, and function [Citation83–85]. However, there are ‘PINK1-PRKN-dependent’ and ‘PINK1-dependent but PRKN-independent’ pathways. It is possible that spautin-1 may function in non-mitophagy and cytosolic functions of PINK1, such as activating AKT or PRKA/PKA [Citation86,Citation87]. Of note, although the different concentrations of spautin-1 have the positive effect on mitophagy (Fig. S1G; Fig. S2E), we did not observe detectable toxicity of spautin-1 in cells (Fig. S1P-S1Q; Fig. S5A-5C) or in C. elegans (Fig.S7A-S7D) in this study. These data indicate that spautin-1 is effective and safe in both cells and animals.
There are several limitations of this study. First, we only used the C. elegans AD model, and have not tested the potential therapeutic effect of spautin-1 in neurodegenerative diseases such as AD and PD in mouse models. The HsMAPT/Tau P301L C. elegans model is under the control of the aex-3 promoter. The aex-3 gene in C. elegans is widely expressed in neurons [Citation88], but some studies also showed that it also functions within the intestinal system [Citation89]. This implies that the expression of MAPT/tau4R1N (P301L) might extend beyond neurons, possibly occurring in other tissues such as the intestine. Nevertheless, the established role of aex-3 in synaptic transmission, which is critical in Alzheimer disease pathology, supports the use of this strain as a model for AD research [Citation61,Citation88]. Further studies in other AD animal models and AD patient iPSC-derived neurons are needed. Moreover, the specificity of RNAi targeting on pink-1 and pdr-1 can be a concern due to sequence similarities with other genes. Our RNAi-based pink-1 and pdr-1 knockdown experiments shall be further validated in mutant animal strains and mice in the future. Second, we mainly tested the effects of spautin-1 on mitophagy using HeLa expressing YFP-PRKN cells and SH-SY5Y cells, not yet in real neuron cells. The metabolism of tumor cells is considerably different from that of neuron cells, as tumor cells predominantly rely on glycolysis with production of lactate instead of mitochondrial oxidation, even in the presence of oxygen [Citation90]. Our data indicate that spautin-1 had little influence on ECAR (), implying that spautin-1 does not affect glycolysis in the cells used in the current study. Thus, the mitophagy promoting effects of spautin-1 could be applicable in neurons given that PINK1-PRKN pathway is essential for mitophagy in neurons and that the promoting effects of spautin-1 is dependent on PINK1. Third, in cell studies, both higher (e.g., 20 µM, 40 µM) and low (1 µM, Fig. S1C, S1G, and Fig. S2E) doses of spautin-1 induced PINK1 stabilization and upregulated mitophagy. On the other hand, the toxicity experiments of worms revealed that higher doses (50 µM, 100 µM, 200 µM) of spautin-1 were detrimental, which inhibited the embryonic development and growth of nematodes (Fig. S7A, S7C). This adverse effect may stem from the inhibitory effect of high-dose spautin-1 on macroautophagy. Hence, for worm experiments, a low dose (1 µM) that induced mitophagy but have only minor effect on macroautophagy was selected. We interpret the dose differences of spautin-1 used in in vitro and in vivo studies through the fundamental physiological disparities between the two systems. Under physiological conditions, the levels of PINK1-PRKN are insufficient for an effective exploration of how spautin-1 regulates PINK1 stability. Nevertheless, when confronted with the non-physiological conditions induced by CCCP, the utilization of higher dose and a short duration in cells allows for a more concentrated examination of spautin-1‘s effect on regulating PINK1 stability. In contrast, in in vivo experiments with AD-like animal models characterized by MAPT/Tau pathology and inherent mitochondrial damage [Citation91], the need for additional CCCP usage in pathological conditions is thus dispensable. Despite the deviation from physiological conditions in cellular experiments, the findings in both models indicate that spautin-1‘s influence on mitophagy is consistently mediated through its effect on PINK1 stability. Hence, it is crucial to acknowledge the difference between each model and to conduct further research to fully justify the effects of spautin-1 on PINK1 stability and mitophagy and its protective effects in AD models. Fourth, despite of the fact that we identified TOMM70 as the main component of the TOMM complex interacting with spautin-1, the functional regions of TOMM70 responsible for this interaction remain to be further investigated. Finally, more work needs to be done to understand the general impact of spautin-1 on the import of other mitochondrial proteins.
In summary, we report a novel function of spautin-1 in promoting mitophagy by stabilization of PINK1 and its potential application in the therapeutic interventions of neurodegenerative diseases.
Materials and Methods
Reagents and antibodies
The following chemical reagents were used in this study: spautin-1 (Sigma-Aldrich, SML0440), carbonyl cyanide 3-chlorophenyl hydrazone (CCCP; Sigma-Aldrich, C2759), MG-132 (Sigma-Aldrich, C2211), oligomycin A (Selleck Chemicals, S1478), antimycin A (Sigma-Aldrich, A8674), valinomycin (Sigma-Aldrich, V3639), torin1 (Sigma-Aldrich, 475,991), bafilomycin A1 (BafA1; Sigma-Aldrich, B1793) and Earle’s Balanced Salts (EBSS; Sigma-Aldrich, E2888), Z-VAD-FMK (Sigma-Aldrich, V116). Amersham ECL prime detection reagent was from GE Healthcare (RPN2232).
Antibodies used in this study: The anti-PINK1 (Cell Signaling Technology, 6946S), anti-TOMM20 (Cell Signaling Technology, 42,406S), anti-MFN2/mitofusin 2 (Cell Signaling Technology, 11,925S), anti-MFN1/mitofusin 1 (Cell Signaling Technology, 14,739S), anti-cleaved CASP3/caspase 3 (Cell Signaling Technology, 9664S), anti-COX4/COXIV (Cell Signaling Technology, 4850S), anti-USP8 (Cell Signaling Technology, 8728S), anti-USP15 (Cell Signaling Technology, 66,310S). The anti-CALCOCO2/NDP52 (Abcam, ab68588), anti-USP10 (Abcam, ab109,219), anti-USP13 (Abcam, ab109,264). The anti-PRKN/parkin (Santa Cruz Biotechnology, sc-32,282), anti-TIMM3 (BD Biosciences, 611,223), anti-phospho-ubiquitin (Ser65) (Merck Millipore, ABS1513-I), anti-DNA (Progen Biotechnik, 61,014), anti-LC3B (Sigma-Aldrich, L7543), anti-GAPDH (Proteintech, 60,004-1-Ig), anti-FLAG (Thermo Fisher Scientific, MA1-91,878), anti-GFP (Thermo Fisher Scientific, MA5-15,256). The anti-TOMM70 (ABclonal, A4349), anti-TOMM40 (ABclonal, A3213).
Secondary antibodies used in this study: Goat anti-mouse IgG (H+L) Alexa Fluor 633 (Thermo fisher scientific, A-21050), goat anti-rabbit IgG (H+L) Alexa Fluor-633 goat anti-rabbit (Thermo fisher scientific, A-21072). Peroxidase-conjugated affinity pure goat antimouse IgG, light chain specific (Jackson Immuno Research, 115–035-174), peroxidase-conjugated IgG fraction monoclonal mouse antirabbit, light chain-specific (Jackson Immuno Research, 211–032-171).
Cell lines and cell culture
HeLa cells stably expressing YFP-PRKN (YPH cells), YPH cells stably expressing mitoKeima (YPH-mt-Keima cells), PINK1-KO HeLa cells, SH-SY5Y cells, GFP-LC3-expressing stable HeLa cells (GFP-LC3 HeLa cells) and HEK293T were cultured in DMEM (HyClone, SH30022.01) supplemented with 10% fetal bovine serum (HyClone, SV30160.03), 100 U/ml penicillin and 100 μg/ml streptomycin (Gibco, 15,140,122). YPH cells and YPH-mt-Keima cells were kind gifts from Dr. Richard Youle (National Institute of Neurological Disorders and Stroke, NIH, USA). The PINK1-KO HeLa cell line was a kind gift from Dr. Kal-Leong Lim (Nanyang Technology University). The SH-SY5Y cell line and 293T were purchased from American Type Culture Collection (CRL-2266, CRL-3216).
Plasmids and siRNA.
siRNA used in this study: siRNA targeting PINK1 (hs.Ri.PINK1.13), USP10 (hs.Ri.USP10.13), USP13 (hs.Ri.USP13.13), USP8 (hs.Ri.USP8.13) and USP15 (hs.Ri.USP15.13) were purchased from Integrated DNA Technologies (https://www.idtdna.com/).
Plasmids used in this study: pcDNA3 PARL-FLAG-CT wild type (Addgene, 13639)
[Citation92] and pcDNA3 PARL-FLAG-CT S277G (Addgene, 13615) [Citation93] were gifts from
Dr. Luca Pellegrini; LentiCRISPRv2 puro (Addgene, 98290) [Citation94] was a gift from
Brett Stringer; pMD2.G (Addgene, 12259) and psPAX2 (Addgene, 12260) were gifts
from Didier Trono. pcDNA-DEST47 PINK1 C-GFP (PINK1-C-GFP) [Citation52] was a gift
from Mark Cookson.
TOMM70 KO cell generation by CRISPR-Cas9.
sgRNA targeting TOMM70 were designed with the online CRISPR design tool (https://www.thermofisher.com/sg/en/home/life-science/genome-editing/crispr-libraries/trueguide-grnas.html). The gRNA sequences (sg-TOMM70-1: 5ʹ-GTGCCATATACCTGTGGAGT-3ʹ, sg-TOMM70-2: 5ʹ-CGCTGCCTCCACAGGTTTAG-3ʹ, sg-TOMM70-3: 5ʹ-TCCGTTCGCTGTTGCGCTTC-3ʹ) were purchased from Sangon Biotech. These sgRNAs were cloned into LentiCRISPRv2 to generate lenti-TOMM70-1, lenti-TOMM70-2 and lenti-TOMM70-3 according to the protocol provided by the Zhang Lab [Citation95]. These plasmids containing the guide RNA of interest and packaging plasmids (PMD2.G and psPAX2; Addgene, 12259 and 12260) were transfected into HEK293T cells. After 96 h, virus supernatant was harvested, and then concentrated using Lentivirus concentration solution (YEASEN, 41101ES50). YPH cells were infected with concentrated virus supernatant for 48 h. Followed by puromycin (400 ng/ml; Merck, P9620) selection for 2 weeks. The TOMM70 knockout effect in puromycin-resistant cells was then verified by immunoblotting.
Immunoblotting
After all designated treatment, cells were rinsed with ice-cold PBS (HyClone, SH30256.01) and lysed with Laemmli buffer (62.5 mM Tris-HCl, pH 6.8, 20% glycerol, 2% SDS, 2 mM dithiothreitol, phosphatase inhibitor and proteinase inhibitor cocktail [Thermo Fisher Scientific, 78,446]). An equal amount of protein was separated on SDS-PAGE gels and then transferred onto PVDF membranes (Bio-Rad Laboratories, 1,620,177). After blocking with 5% nonfat milk (Bio-Rad Laboratories, 1,706,404) for 1 h, incubation with primary antibodies was performed overnight at 4℃. After that, the membranes were incubated with a secondary antibody for 1 h at room temperature. The results were visualized by the Image Quant LAS 500 (GE Healthcare) with an ECL chemiluminescence detection kit (Thermo Fisher Scientific, 34,076).
Immunoprecipitation (IP)
The cells were harvested and lysed in the IP lysis buffer (10 mM Tris-HCl, pH 7.4, 100 mM NaCl, 2.5 mM MgCl2, 0.5% Triton X-100 (Sigma-Aldrich, T8787), phosphatase inhibitor and proteinase inhibitor cocktail). After sonication, the lysate was centrifuged at 12,000 g for 20 min at 4℃. The resultant supernatant was incubated with either IgG or indicated antibodies overnight at 4℃ with gentle rotation, and was subsequently incubated with 10 μl protein A/G (Thermo Fisher Scientific, 20,421) for an additional 4 h. For GFP IP, the supernatant was incubated with 10 μl anti- GFP-Trap Agarose (Proteintech, 17,383,343) overnight at 4℃ with gentle rotation. All the immunoprecipitates were washed with IP lysis buffer 3 times, and then eluted by boiling for 5 min with 1X LDS sample buffer and analyzed with immunoblotting.
Isolation of the mitochondrial fraction
The designated cells were washed with ice-cold PBS twice and detached by scrapping on ice with mitochondrial isolation buffer (200 mM mannitol, 50 mM KCl, 5 mM EGTA, 50 mM HEPES-KOH, pH 7.4, 68 mM sucrose, 2 mM MgCl2, 1 mM dithiothreitol, phosphatase inhibitor and proteinase inhibitor cocktail). Cell suspensions were transferred to tubes and incubated on ice for 20 min. After incubation, the cells were homogenized using a dounce homogenizer with 30-40 up-and-down passes of the pestle followed by centrifugation at 800 g for 10 min at 4℃. The resulting supernatant was collected and centrifuged at 12000 g for 30 min at 4℃. And then, the supernatant was collected as cytosolic fraction, and the mitochondrial pellet was rinsed twice in isolation buffer at 12,000 g for 15 min at 4℃ to yield the crude mitochondrial fraction.
Immunofluorescence microscopy
For PRKN recruitment to mitochondria analyses, post-treatment cells were incubated with MitoTrackerTM Red FM (Thermo Fisher Scientific, M22425) at 37°C in growth media for 15 min. The images were captured with Leica DMi8 fluorescence microscope and percentage of cells with YFP-PRKN mitochondrial translocation was quantified from a minimum of 200 cells using ImageJ software. To measure activity of PINK1 and mitophagy by immunostaining, cells were seeded to a cover glass slide at the confluency of approximately 60%. After designated treatment, cells were fixed for 15 min at room temperature with 4% paraformaldehyde (PFA; Santa Cruz Biotechnology, sc-281,692), and then permeabilized with 0.25% Triton X-100 for 5 min at room temperature. Then, cells were blocked with 1% BSA (Sigma-Aldrich, A2153) for 1 h at room temperature. For immunostaining, cells were incubated with specific primary antibodies overnight at 4°C. In the next day, the antibodies were removed and cells were rinsed with 0.1% Tween-20 (Sigma-Aldrich, P9416) in PBS (PBST) followed by incubation with anti-rabbit or anti-mouse Alexa Fluor 633-conjugated secondary antibody for 1 h at room temperature. After that, cells were rinsed again with PBST three times and the cover glass slides were sealed with mounting medium containing DAPI (Thermo Fisher Scientific, P36980). The images were obtained with a confocal microscope (Olympus Fluoview FV3000, Olympus America Inc., PA) or Leica DMi8 fluorescence microscope. To measure mitophagy by mtDNA, the level of mtDNA was quantified following the well-established methodology described previously, including in a few of our own published works [Citation40,Citation96–98]. Briefly, Imaris 9.1 software was used to analyze a minimum of 200 cells in three independent experiments. We used the following formula to calculated the stain remaining: (cDNAv-nDNAv)/n (cDNAv representing the total cellular DNA volume determined by staining using anti-DNA antibodies; nDNAv representing the volume of the anti-DNA localized in the nuclear DNA (nDNA) by staining using DAPI; n representing the number of cells). The mtDNA stain volume in DMSO group was normalized to 100%.
Real-time quantitative PCR (qPCR) analysis of mitochondrial DNA (mtDNA) content
For the measurement of mtDNA content, we followed the methodology described previously [Citation63,Citation99]. Briefly, the cellular DNA were first extracted from the treated YPH cells using the DNeasy blood and tissue kit (Qiagen, 69506). Then, mitochondrial MT-RNR2/16S RNA gene or B2M/β2-microglobulin gene were utilized as mtDNA or nDNA marker, respectively, and their levels were quantified by a CFX96 real-time PCR detection system (Bio-Rad) with Hieff® qPCR SYBR Green Master Mix (Yeasen, 11202ES08) using the follow primers: MT-RNR2/16S rRNA gene (forward, 5-GCCTTCCCCCGTAAATGATA-3; reverse, 5-TTATGCGATTACCGGGCTCT-3) and B2M gene (forward, 5 -TGCTGTCTCCATGTTTGATGTATCT-3; reverse, 5-TCTCTGCTCCCCACCTCTAAGT-3).
Measurement of mitochondrial membrane potential (MMP)
After designated treatments, cells were incubated at 37°C with TMRE (100 nM) (Thermo Fisher Scientific, I 34361) in growth media for 30 min in a CO2 incubator. After incubation, cells were analyzed using fluorescence microscopy or flow cytometery to determine the fluorescence intensity. Measurement of TMRE integrated intensity was done using ImageJ.
Cell Counting Kit-8 (CCK-8) cell viability assay
YPH cells (5×103) cells were planted in 96-well plates. After cells were treated with different concentrations of spautin-1 for 18 h, 10 μl CCK-8 (TransGen, FC101-01) and 90 ul medium were incubated together at 37℃ for 1 h, then the absorbance at 450 nm was measured by a microplate reader (Bio-Rad, Hercules, CA, USA).
Oxygen consumption rate (OCR) and extracellular acidification rate (ECAR)
OCR and ECAR measurements using seahorse cellular flux assays. Seahorse plates were pretreated by coating with poly-d-lysine (0.1 mg/ml) and Matrigel (Sigma-Aldrich, E0282). Cells were plated with 5 × 103 cells per XF96 well to ensure ~90% surface coverage at the time of the experiment the next day. For OCR analysis, medium was exchanged for mitochondrial stress medium (Seahorse XF Assay Medium supplemented with 2 mM glutamine and 10 mM glucose) at 1 hour before the assay. The cultures were similarly equilibrated in non-CO2 incubator. Substrates and selective inhibitors were injected to achieve final concentrations of oligomycin (Sigma-Aldrich, 495455) at 1.0 μM, carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone (FCCP; Sigma-Aldrich, C2920) at 0.5 μM, and rotenone-antimycin A (Sigma-Aldrich, 557368 and A8674,) at 0.5 μM, according to the manufacturer’s instructions. For glycolysis analyses, medium was exchanged for Seahorse Glycolysis Stress medium (Seahorse XF Assay Medium supplemented with 2 mM glutamine) at 1 h before the assay. The cultures were similarly equilibrated in non-CO2 incubator. Substrates and selective inhibitors were injected to achieve final concentrations of glucose at 10 mM, oligomycin at 1.5 μM, and 2-deoxy-D-glucose (2-DG; Medchemexpress, HY-13966) at 50 mM, according to the manufacturer’s instructions. The OCR and ECAR values were further normalized to the number of cells present in each well.
Molecular docking between spautin-1 and target proteins
The crystal structures of candidate proteins were downloaded from the RCSB Protein DataBank (http://www.rcsb.org/). ChemBioOffice 2014 was used to create 3D chemical structure and minimize its energy. Employing AUTODOCK VINA 1.1.2 software generated the molecular docking model, and the docking score between the compound and the target proteins was presented as kcal/mol and used as the evaluation criteria. Targets with docking scores < -6 were considered meaningful [Citation100].
Streptavidin-biotin affinity affinity-isolation assay
Biotin-spautin-1 (bio-spautin-1) were synthesized as described in Supporting Information. YPH cells were lysed with IP lysis buffer containing 1% protease inhibitor cocktail. After sonication, cell lysates were centrifuged at 12,000 g for 20 min at 4℃. The supernatant was incubated with free biotin (Sigma-Aldrich, B4501) or bio-spautin-1 (50 μM) for 4 h at 4°C with gently rotation. Subsequently, the pre-washed streptavidin magnetic beads (Thermo Fisher Scientific, 88816) were added to the system as above and incubated overnight at 4°C with gentle rotation. The beads were washed three times with elution buffer then the precipitate was separated by SDS-PAGE and analyzed by immunoblotting.
C.elegans strains, genetics and maintenance
A list of strains used in this study is shown in Table S1. Escherichia coli (OP50; LabTIE’s Freeze-dried OP50) at a concentration of 1 × 1011 cfu/mL was used as a food source for the maintenance of all the C. elegans strains at 20°C on standard nematode growth medium (NGM) plates [Citation101,Citation102]. Unless otherwise stated, all experiments were conducted at 20°C. For each strain, adult day 2 worms (via a 6-h egg lay method, and wait two days after the L4 stage) were isolated and then bleached to obtain a synchronized population of worms [Citation103]. To avoid effects of early-stage starvation, egg-containing M9 buffer (22 mM KH2PO3, 42.3 mM Na2HPO3, pH 7.4, 85.5 mM NaCl, 0.1% MgSO4) was pipetted onto designated OP50-containing NGM plates and evenly spread [Citation102]. Eggs were then plated and allowed to grow until they developed into day 2 of adulthood. The compound DMSO-dissolved spautin-1 was added in NGM shortly before plating. The spautin-1 was administered to worms either at egg hatching or the L4 stage. Specifically, from egg hatching until the L4 stage is 48 h, and from the L4 stage until adult day 2 requires an additional 48 h, making the total exposure time to Spautin-1 either 48 or 96 h.
RNAi knockdown in C. elegans
In C. elegans, sid-1 (systemic RNA interference-deficient) is a gene that plays a crucial role in the uptake of double-stranded RNA (dsRNA) from the environment [Citation104]. The neuronal overexpression of sid-1 (n-sid1OV) in C. elegans involves increasing the expression of the sid-1 gene specifically in neurons [Citation105,Citation106]. By using n-sid1OV strains, the spread of RNA interference (RNAi) effects across the nervous system can be modulated [Citation105]. Four neuronal over-expression of sid-1 strains were used to carry out RNAi knockdown of designated genes (pink-1 or pdr-1 or lgg-1). The dsRNA-expressing bacteria were grown on Luria-Bertani (LB) agar plates supplemented with ampicillin at 37°C overnight, followed by inoculation of a bacterial colony into LB broth supplemented with ampicillin and overnight growth. Prepared bacterial cultures were inoculated onto 1-day-old NGM plates containing 1 mM IPTG (Millipore, 420322) and 100 µg/ml ampicillin and allowed to dry for 24 or 48 h [Citation107]. Synchronized embryos obtained by bleaching were added to each dried plate and incubated until day 2 of adulthood.
Evaluation of neuronal mitophagy inducers using C. elegans mitophagy reporter strains
Neuronal mt-Rosella reporter strain (neuronal mt-Rosella: Ex[punc-119TOMM-20:Rosella;rol-6(su1006)]) has been used to quantify neuronal mitophagy in worms. Transgenic worms expressing a pan-neuronal mt-Rosella biosensor using a GFP variant that was acid-sensitive in the lysosomal lumen and was fused with the pH-insensitive Discosoma red fluorescent protein (DsRed), as described elsewhere [Citation48,Citation61,Citation62]. GFP:DsRed was used to measure mitophagy. Therefore, mitophagy was elevated at lower pixel intensity ratios. In this study, mt-Rosella-expressing nematodes (N2;mt-Rosella), a neuronal over-expression of sid-1 strain [Citation105] with mt-Rosella biosensor (N2n-sid-1OV;mt-Rosella), and a neuronal over-expression of sid-1 in AD-like HsMAPT/TauP301L with mt-Rosella (HsMAPT/TauP301L;n-sid-1OV;mt-Rosella) strains were used. Here nematodes were given 1 μM of spautin-1 and the mitophagy level was assessed on the 2-day-old adult nematodes. CCCP was used as a positive control to trigger mitophagy.
C.elegans chemotaxis assay
Chemotaxis assay in C. elegans is commonly used to study the ability of worms to move toward or away from chemical cues in their environment. It is associated with associative learning capabilities in C. elegans. As described previously, chemotaxis to soluble and volatile compounds (isoamyl alcohol, IA) was investigated [Citation48,Citation61,Citation108]. As previously developed [Citation102], the current study adopted the following methodology. Firstly, synchronized worms (around 200 worms per group) were collected and washed with M9 buffer to remove residual bacteria, followed by placement in plain 6-cm NGM plates (with no OP50) with/without IA for 90 min. For naïve plates, seal plates with parafilm, and place the plates upside down until next step. For IA plates, a droplet of 10 μl IA was dropped onto the middle of the lid of the IA conditioning plates. Ten-centimeter assay plates were then prepared by adding 20 μl 20 mM NaN3 to the ‘IA’ and ‘T’ points, respectively. After drying the assay plates (20-22°C) for 30 min, tests were run. Each plate was covered with 50 x 50 mm Parafilm over the ‘IA’ area. The worms were collected and washed with M9 buffer following the conditioning step. Then, the washed worms were placed on the ‘S’ point area. On the ‘IA’ area, 5 μl of 2% IA were applied to the Parafilm. The worms were then left at room temperature for 2 h. After that, the number of worms in the ‘S’, ‘IA’, and ‘T’ regions was determined. A chemotaxis index was established as: (#‘IA’ - #‘T’/(#‘IA’ + #‘T’ + #‘S’), with ‘#’ denoting numbers [Citation61,Citation102,Citation109]. A lower chemotaxis index is associated with better performance. Biological replications were performed three times. All chemotaxis assays were conducted using adult day 2 worms.
C.elegans lifespan assay
A lifespan evaluation was conducted according to established protocols [Citation61,Citation110] with minor modifications, using 3.5 cm NGM plates that were pre-seeded with E. coli OP50 three days before the experiment. Spautin-1 was introduced during the pouring of the NGM plates. The conception of synchronized animal populations was achieved through a 6-h egg laying by gravid adults at day 2, followed by precisely defined conditions for embryonic development into adulthood. For each condition, 20 worms at the L4 stage (day 0) were transferred to 3.5 cm plates to establish synchronous populations of 120 animals. A survival status was monitored daily, and animals were transferred every two days to fresh OP50 seeded plates (-/+ spautin-1) during the fertile period, and subsequently every 2 days until day 8 of adulthood. When pharyngeal pumping ceased and worms were unresponsive to touch, worms were considered dead. Censored data included worms that died due to internal bagging, crawling on the plate edge, or gonad extrusion. For mortality calculations, these animals were given a weight of ‘0’ up to the point of censorship in the lifespan analysis.
C.elegans toxicity assay
Toxicity experiments were conducted on N2 C. elegans at 20°C, including fecundity (3-h egg-laying), hatching, and larval development [Citation58,Citation111]. In brief, ten adult day 2 worms were transferred onto OP50 seeded NGM plates containing 0.5% DMSO or spautin-1 (0, 1, 10, 50, 100, 200 μM). Following a 3-h egg-laying period, gravid worms were removed from the plates. The reproductivity of the worms was then determined based on the number of eggs laid by the worms. A hatching efficiency test was carried out 12 h of incubation to count the number of hatched eggs and L1 larvae. After the 48-h period following egg laying, development was evaluated by measuring the number of L4 larvae. As a final step, the growth of L4 larvae to adulthood was measured 16 h after the L4 larval stage. A total of 30 worms were placed on three plates in each group.
Statistics analysis
The statistical significance of the mean differences observed between two samples was determined by Student’s t test, and One-way ANOVA or Brown-Forsythe and Welch ANOVA tests followed by Multiple comparisons test using GraphPad Prism 8. Data are shown as means ± S.D. of the results of at least 3 independent experiments. Values of P < 0.05 were considered significant.
Declaration of interests
E.F.F. has an MTA with LMITO Therapeutics Inc (South Korea), a CRADA arrangement with ChromaDex (USA), and a commercialization agreement with Molecule AG/VITADAO, and is a consultant to Aladdin Healthcare Technologies (UK and Germany), the Vancouver Dementia Prevention Centre (Canada), Intellectual Labs (Norway), and Mind Rank AI.
Upon mitochondrial damage, stabilization of PINK1 by spautin-1 promotes PINK1-PRKN-mediated mitophagy. This process is initiated through direct bound with TOMM complex responsible for stability of PINK1. Eventually, spautin-1 restores deficient mitophagy to improve associative learning capability in MAPT/Tau AD C. elegans model.
Supplementary materials and figures R6.docx
Download MS Word (1.8 MB)Acknowledgements
We thank members of the Shen laboratory for valuable discussion. We thank Dr. Kal-Leong Lim from Nanyang Technology University, Singapore, for his critical comments. We thank Dr. Richard Youle for providing YPH cells and YPH-mt-Keima cells.
Supplementary material
Supplemental data for this article can be accessed here
Additional information
Funding
References
- Dikic I Proteasomal and Autophagic Degradation Systems. Annu Rev Biochem. 2017 Jun 20;86:193–224. 1 10.1146/annurev-biochem-061516-044908
- Lu G, Wang Y, Shi Y, et al. Autophagy in health and disease: From molecular mechanisms to therapeutic target. MedComm (2020). 2022Sep;3(3):e150.
- Yamamoto H, Zhang S, Mizushima N Autophagy genes in biology and disease. Nat Rev Genet. 2023 Jun;24(6):382–400.10.1038/s41576-022-00562-w
- Wu Y, Tan HWS, Lin J-Y, et al. Molecular mechanisms of autophagy and implications in liver diseases. Liver Research. 20232023/03 /01/;7(1):56–70.
- Tracy TE, Madero-Pérez J, Swaney DL, et al. Tau interactome maps synaptic and mitochondrial processes associated with neurodegeneration. Cell. 2022 Feb 17;185(4):712–728.e14.
- Zachari M, Ganley IG, Lane JD, et al. The mammalian ULK1 complex and autophagy initiation. Essays Biochem. 2017 Dec 12;61(6):585–596. 10.1042/EBC20170021
- Gao M, Monian P, Pan Q, et al. Ferroptosis is an autophagic cell death process. Cell Res. 2016 Sep;26(9):1021–1032.10.1038/cr.2016.95
- Mizushima N The ATG conjugation systems in autophagy. Curr Opin Cell Biol. 2020 Apr;63:1–10. 10.1016/j.ceb.2019.12.001
- Diao J, Liu R, Rong Y, et al. ATG14 promotes membrane tethering and fusion of autophagosomes to endolysosomes. Nature. 2015 Apr 23;520(7548):563–566.10.1038/nature14147
- Shi Q, Pei F, Silverman GA, et al. Mechanisms of Action of Autophagy Modulators Dissected by Quantitative Systems Pharmacology Analysis. Int J Mol Sci. 2020 Apr 19;21(8). 2855 10.3390/ijms21082855
- Vakifahmetoglu-Norberg H, Xia HG, Yuan J Pharmacologic agents targeting autophagy. J Clin Invest. 2015 Jan;125(1):5–13. 10.1172/JCI73937
- Liu J, Xia H, Kim M, et al. Beclin1 controls the levels of p53 by regulating the deubiquitination activity of USP10 and USP13. Cell. 2011 Sep 30;147(1):223–234.10.1016/j.cell.2011.08.037
- Vakifahmetoglu-Norberg H, Kim M, Xia HG, et al. Chaperone-mediated autophagy degrades mutant p53. Genes Dev. 2013 Aug 1;27(15):1718–1730. 10.1101/gad.220897.113
- Elsocht M, Giron P, De Grève J, et al. Second generation Spautin-1 analogues targeting EGFR-mutant non-small cell lung cancer cells. Bioorg Med Chem Lett. 2023 Jan 1;79:129066. 10.1016/j.bmcl.2022.129066
- Elsocht M, Giron P, Maes L, et al. Structure-Activity Relationship (SAR) Study of Spautin-1 to Entail the Discovery of Novel NEK4 Inhibitors. Int J Mol Sci. 2021 Jan 10;22(2).
- Kunimasa K, Ikeda-Ishikawa C, Tani Y, et al. Spautin-1 inhibits mitochondrial complex I and leads to suppression of the unfolded protein response and cell survival during glucose starvation. Sci Rep. 2022 Jul 7;12(1):11533.10.1038/s41598-022-15673-x
- Nunnari J, Suomalainen A Mitochondria: in sickness and in health. Cell. 2012 Mar 16;148(6):1145–1159.10.1016/j.cell.2012.02.035
- Shen K, Pender CL, Bar-Ziv R, et al. Mitochondria as Cellular and Organismal Signaling Hubs. Annu Rev Cell Dev Biol. 2022 Oct 6;38:179–218. 1 10.1146/annurev-cellbio-120420-015303
- Bao F, Xiao J, Zhou L, et al.. Autophagy-independent mitochondrial quality control: Mechanisms and disease associations. MedComm – Future Medicine. 2022;1(2):e25. doi: 10.1002/mef2.25
- Spinelli JB, Haigis MC The multifaceted contributions of mitochondria to cellular metabolism. Nat Cell Biol. 2018 Jul;20(7):745–754.10.1038/s41556-018-0124-1
- Trigo D, Avelar C, Fernandes M, et al. Mitochondria, energy, and metabolism in neuronal health and disease. FEBS Lett. 2022 May;596(9):1095–1110. 10.1002/1873-3468.14298
- Wang Y, Liu HH, Cao YT, et al. The Role of Mitochondrial Dynamics and Mitophagy in Carcinogenesis, Metastasis and Therapy. Front Cell Dev Biol. 2020;8:413. 10.3389/fcell.2020.00413
- Youle RJ, Narendra DP Mechanisms of mitophagy. Nat Rev Mol Cell Biol. 2011 Jan;12(1):9–14.10.1038/nrm3028
- Youle RJ, van der Bliek AM Mitochondrial fission, fusion, and stress. Science. 2012 Aug 31;337(6098):1062–1065.10.1126/science.1219855
- Deas E, Plun-Favreau H, Gandhi S, et al. PINK1 cleavage at position A103 by the mitochondrial protease PARL. Hum Mol Genet. 2011 Mar 1;20(5):867–879. 10.1093/hmg/ddq526
- Jin SM, Lazarou M, Wang C, et al. Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. J Cell Biol. 2010 Nov 29;191(5):933–942. 10.1083/jcb.201008084
- Maruszczak KK, Jung M, Rasool S, et al. The role of the individual TOM subunits in the association of PINK1 with depolarized mitochondria. J Mol Med (Berl). 2022 May;100(5):747–762.10.1007/s00109-022-02191-6
- Meissner C, Lorenz H, Weihofen A, et al. The mitochondrial intramembrane protease PARL cleaves human Pink1 to regulate Pink1 trafficking. J Neurochem. 2011;117(5):856–867. doi: 10.1111/j.1471-4159.2011.07253.x
- Yamano K, Youle RJ PINK1 is degraded through the N-end rule pathway. Autophagy. 2013 Nov 1;9(11):1758–1769.10.4161/auto.24633
- Callegari S, Cruz-Zaragoza LD, Rehling P. From TOM to the TIM23 complex - handing over of a precursor. Biol Chem. 2020 May 26;401(6–7):709–721.
- Lazarou M, Jin SM, Kane LA, et al. Role of PINK1 binding to the TOM complex and alternate intracellular membranes in recruitment and activation of the E3 ligase Parkin. Dev Cell. 2012 Feb 14;22(2):320–333.10.1016/j.devcel.2011.12.014
- Okatsu K, Uno M, Koyano F, et al. A dimeric PINK1-containing complex on depolarized mitochondria stimulates Parkin recruitment. J Biol Chem. 2013 Dec 20;288(51):36372–36384. 10.1074/jbc.M113.509653
- Rasool S, Veyron S, Soya N, et al. Mechanism of PINK1 activation by autophosphorylation and insights into assembly on the TOM complex. Mol Cell. 2022 Jan 6;82(1):44–59.e6.
- Caulfield TR, Fiesel FC, Moussaud-Lamodière EL, et al. Phosphorylation by PINK1 releases the UBL domain and initializes the conformational opening of the E3 ubiquitin ligase Parkin. PLoS Comput Biol. 2014 Nov;10(11):e1003935. 10.1371/journal.pcbi.1003935
- Kane LA, Lazarou M, Fogel AI, et al. PINK1 phosphorylates ubiquitin to activate Parkin E3 ubiquitin ligase activity. J Cell Biol. 2014 Apr 28;205(2):143–153. 10.1083/jcb.201402104
- Kondapalli C, Kazlauskaite A, Zhang N, et al. PINK1 is activated by mitochondrial membrane potential depolarization and stimulates Parkin E3 ligase activity by phosphorylating Serine 65. Open Biol. 2012 May;2(5):120080. 10.1098/rsob.120080
- Ordureau A, Sarraf SA, Duda DM, et al. Quantitative proteomics reveal a feedforward mechanism for mitochondrial PARKIN translocation and ubiquitin chain synthesis. Mol Cell. 2014 Nov 6;56(3):360–375.10.1016/j.molcel.2014.09.007
- Shiba-Fukushima K, Imai Y, Yoshida S, et al. PINK1-mediated phosphorylation of the parkin ubiquitin-like domain primes mitochondrial translocation of parkin and regulates mitophagy. Sci Rep. 2012;2(1):1002. doi: 10.1038/srep01002
- Heo JM, Ordureau A, Paulo JA, et al. The PINK1-PARKIN Mitochondrial Ubiquitylation Pathway Drives a Program of OPTN/NDP52 Recruitment and TBK1 Activation to Promote Mitophagy. Mol Cell. 2015 Oct 1;60(1):7–20.10.1016/j.molcel.2015.08.016
- Lazarou M, Sliter DA, Kane LA, et al. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature. 2015 Aug 20;524(7565):309–314.10.1038/nature14893
- Sarraf SA, Raman M, Guarani-Pereira V, et al. Landscape of the PARKIN-dependent ubiquitylome in response to mitochondrial depolarization. Nature. 2013 Apr 18;496(7445):372–376.10.1038/nature12043
- Wong YC, Holzbaur EL. Optineurin is an autophagy receptor for damaged mitochondria in parkin-mediated mitophagy that is disrupted by an ALS-linked mutation. Proc Natl Acad Sci U S A. 2014 Oct 21;111(42):E4439–48.
- Heo JM, Harper NJ, Paulo JA, et al. Integrated proteogenetic analysis reveals the landscape of a mitochondrial-autophagosome synapse during PARK2-dependent mitophagy. Sci Adv. 2019 Nov;5(11):eaay4624. 10.1126/sciadv.aay4624
- Vargas JNS, Wang C, Bunker E, et al. Spatiotemporal Control of ULK1 Activation by NDP52 and TBK1 during Selective Autophagy. Mol Cell. 2019 Apr 18;74(2):347–362.e6.
- Nguyen TN, Sawa-Makarska J, Khuu G, et al. Unconventional initiation of PINK1/Parkin mitophagy by Optineurin. Mol Cell. 2023 May 18;83(10):1693–1709.e9.
- Sun N, Youle RJ, Finkel T The Mitochondrial Basis of Aging. Mol Cell. 2016 Mar 3;61(5):654–666.10.1016/j.molcel.2016.01.028
- Chin RM, Rakhit R, Ditsworth D, et al. Pharmacological PINK1 activation ameliorates Pathology in Parkinson’s Disease models. bioRxiv. 2023 Feb 15.
- Fang EF, Hou Y, Palikaras K, et al. Mitophagy inhibits amyloid-β and tau pathology and reverses cognitive deficits in models of Alzheimer’s disease. Nat Neurosci. 2019 Mar;22(3):401–412.10.1038/s41593-018-0332-9
- Kerr JS, Adriaanse BA, Greig NH, et al. Mitophagy and Alzheimer’s Disease: Cellular and Molecular Mechanisms. Trends Neurosci. 2017 Mar;40(3):151–166. 10.1016/j.tins.2017.01.002
- Sliter DA, Martinez J, Hao L, et al. Parkin and PINK1 mitigate STING-induced inflammation. Nature. 2018 Sep;561(7722):258–262.10.1038/s41586-018-0448-9
- Lautrup S, Sinclair DA, Mattson MP, et al. NAD(+) in Brain Aging and Neurodegenerative Disorders. Cell Metab. 2019 Oct 1;30(4):630–655.
- Beilina A, Van Der Brug M, Ahmad R, et al. Mutations in PTEN-induced putative kinase 1 associated with recessive parkinsonism have differential effects on protein stability. Proc Natl Acad Sci U S A. 2005 Apr 19;102(16):5703–5708. 10.1073/pnas.0500617102
- Du H, Guo L, Fang F, et al. Cyclophilin D deficiency attenuates mitochondrial and neuronal perturbation and ameliorates learning and memory in Alzheimer’s disease. Nat Med. 2008 Oct;14(10):1097–1105.10.1038/nm.1868
- Lustbader JW, Cirilli M, Lin C, et al. ABAD directly links Abeta to mitochondrial toxicity in Alzheimer’s disease. Science. 2004 Apr 16;304(5669):448–52.
- Manczak M, Calkins MJ, Reddy PH Impaired mitochondrial dynamics and abnormal interaction of amyloid beta with mitochondrial protein Drp1 in neurons from patients with Alzheimer’s disease: implications for neuronal damage. Hum Mol Genet. 2011 Jul 1;20(13):2495–2509.10.1093/hmg/ddr139
- Hu Y, Li XC, Wang ZH, et al. Tau accumulation impairs mitophagy via increasing mitochondrial membrane potential and reducing mitochondrial Parkin. Oncotarget. 2016 Apr 5;7(14):17356–17368.10.18632/oncotarget.7861
- Li XC, Hu Y, Wang ZH, et al. Human wild-type full-length tau accumulation disrupts mitochondrial dynamics and the functions via increasing mitofusins. Sci Rep. 2016 Apr 21;6:24756. 1 10.1038/srep24756
- Chen C, Yang C, Wang J, et al. Melatonin ameliorates cognitive deficits through improving mitophagy in a mouse model of Alzheimer’s disease. J Pineal Res. 2021 Dec;71(4):e12774. 10.1111/jpi.12774
- Du F, Yu Q, Yan S, et al. PINK1 signalling rescues amyloid pathology and mitochondrial dysfunction in Alzheimer’s disease. Brain. 2017 Dec 1;140(12):3233–3251.10.1093/brain/awx258
- Lou G, Palikaras K, Lautrup S, et al. Mitophagy and Neuroprotection. Trends Mol Med. 2020 Jan;26(1):8–20. 10.1016/j.molmed.2019.07.002
- Xie C, Zhuang XX, Niu Z, et al. Amelioration of Alzheimer’s disease pathology by mitophagy inducers identified via machine learning and a cross-species workflow. Nat Biomed Eng. 2022 Jan;6(1):76–93.
- Fang EF, Palikaras K, Sun N, et al. In Vitro and In Vivo Detection of Mitophagy in Human Cells, C. Elegans, and Mice. J Vis Exp. 2017 Nov 22(129).
- Amadoro G, Corsetti V, Florenzano F, et al. AD-linked, toxic NH2 human tau affects the quality control of mitochondria in neurons. Neurobiol Dis. 2014 Feb;62:489–507. 10.1016/j.nbd.2013.10.018
- Lazarou M, Narendra DP, Jin SM, et al. PINK1 drives Parkin self-association and HECT-like E3 activity upstream of mitochondrial binding. J Cell Biol. 2013 Jan 21;200(2):163–172. 10.1083/jcb.201210111
- Bingol B, Tea JS, Phu L, et al. The mitochondrial deubiquitinase USP30 opposes parkin-mediated mitophagy. Nature. 2014 Jun 19;510(7505):370–375.10.1038/nature13418
- Cornelissen T, Haddad D, Wauters F, et al. The deubiquitinase USP15 antagonizes Parkin-mediated mitochondrial ubiquitination and mitophagy. Hum Mol Genet. 2014 Oct 1;23(19):5227–5242. 10.1093/hmg/ddu244
- Durcan TM, Tang MY, Pérusse JR, et al. USP8 regulates mitophagy by removing K6-linked ubiquitin conjugates from parkin. EMBO J. 2014 Nov 3;33(21):2473–2491. 10.15252/embj.201489729
- Kazlauskaite A, Kelly V, Johnson C, et al. Phosphorylation of Parkin at Serine65 is essential for activation: elaboration of a Miro1 substrate-based assay of Parkin E3 ligase activity. Open Biol. 2014 Mar 19;4(3):130213. 10.1098/rsob.130213
- Koyano F, Okatsu K, Kosako H, et al. Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature. 2014 Jun 5;510(7503):162–166.10.1038/nature13392
- Sha D, Chin LS, Li L. Phosphorylation of parkin by Parkinson disease-linked kinase PINK1 activates parkin E3 ligase function and NF-kappaB signaling. Hum Mol Genet. 2010 Jan 15;19(2):352–63.
- Li J, Fu A, Zhang L. An Overview of Scoring Functions Used for Protein-Ligand Interactions in Molecular Docking. Interdiscip Sci. 2019 Jun;11(2):320–328.
- Kato H, Lu Q, Rapaport D, et al. Tom70 is essential for PINK1 import into mitochondria. PLoS One. 2013;8(3):e58435. doi: 10.1371/journal.pone.0058435
- Pickles S, Vigié P, Youle RJ Mitophagy and Quality Control Mechanisms in Mitochondrial Maintenance. Curr Biol. 2018 Feb 19;28(4):R170–r185. 10.1016/j.cub.2018.01.004
- Schmidt O, Pfanner N, Meisinger C Mitochondrial protein import: from proteomics to functional mechanisms. Nat Rev Mol Cell Biol. 2010 Sep;11(9):655–667.10.1038/nrm2959
- Kreimendahl S, Rassow J The Mitochondrial Outer Membrane Protein Tom70-Mediator in Protein Traffic, Membrane Contact Sites and Innate Immunity. Int J Mol Sci. 2020 Oct 1;21(19). 7262 10.3390/ijms21197262
- Schleiff E, Turnbull JL Functional and structural properties of the mitochondrial outer membrane receptor Tom20. Biochemistry. 1998 Sep 22;37(38):13043–13051.10.1021/bi9807456
- Wu Y, Sha B Crystal structure of yeast mitochondrial outer membrane translocon member Tom70p. Nat Struct Mol Biol. 2006 Jul;13(7):589–593.10.1038/nsmb1106
- Kingwell K Turning up mitophagy in Alzheimer disease. Nat Rev Drug Discov. 2019 Mar 4.10.1038/d41573-019-00035-6
- Grishchuk Y, Ginet V, Truttmann AC, et al. Beclin 1-independent autophagy contributes to apoptosis in cortical neurons. Autophagy. 2011 Oct;7(10):1115–1131.10.4161/auto.7.10.16608
- Scarlatti F, Maffei R, Beau I, et al. Role of non-canonical Beclin 1-independent autophagy in cell death induced by resveratrol in human breast cancer cells. Cell Death Differ. 2008 Aug;15(8):1318–1329.10.1038/cdd.2008.51
- Wang Y, Yue Y, Sun YH, et al. Can bispectral index or auditory evoked potential index predict implicit memory during propofol-induced sedation? Chin Med J (Engl). 2006 Jun 5;119(11):894–898. 10.1097/00029330-200606010-00003
- Fu H, Possenti A, Freer R, et al. A tau homeostasis signature is linked with the cellular and regional vulnerability of excitatory neurons to tau pathology. Nat Neurosci. 2019 Jan;22(1):47–56.10.1038/s41593-018-0298-7
- Fallon L, Moreau F, Croft BG, et al. Parkin and CASK/LIN-2 associate via a PDZ-mediated interaction and are co-localized in lipid rafts and postsynaptic densities in brain. J Biol Chem. 2002 Jan 4;277(1):486–491. 10.1074/jbc.M109806200
- Lieberman OJ, Sulzer D The Synaptic Autophagy Cycle. J Mol Biol. 2020 Apr 3;432(8):2589–2604. 10.1016/j.jmb.2019.12.028
- Otero PA, Fricklas G, Nigam A, et al. Endogenous PTEN-Induced Kinase 1 Regulates Dendritic Architecture and Spinogenesis. J Neurosci. 2022 Oct 12;42(41):7848–7860. 10.1523/JNEUROSCI.0785-22.2022
- Furlong RM, Lindsay A, Anderson KE, et al. The Parkinson’s disease gene PINK1 activates Akt via PINK1 kinase-dependent regulation of the phospholipid PI(3,4,5)P(3).J Cell Sci. 2019 Oct 22;132(20).
- Wang KZQ, Steer E, Otero PA, et al. PINK1 Interacts with VCP/p97 and Activates PKA to Promote NSFL1C/p47 Phosphorylation and Dendritic Arborization in Neurons. eNeuro. 2018 Nov-Dec;5(6). ENEURO.0466-18.2018 10.1523/ENEURO.0466-18.2018
- Iwasaki K, Staunton J, Saifee O, et al. aex-3 encodes a novel regulator of presynaptic activity in C. elegans. Neuron. 1997 Apr;18(4):613–622.10.1016/S0896-6273(00)80302-5
- Mahoney TR, Luo S, Round EK, et al. Intestinal signaling to GABAergic neurons regulates a rhythmic behavior in Caenorhabditis elegans. Proc Natl Acad Sci U S A. 2008 Oct 21;105(42):16350–16355. 10.1073/pnas.0803617105
- Chelakkot C, Chelakkot VS, Shin Y, et al. Modulating Glycolysis to Improve Cancer Therapy. Int J Mol Sci. 2023 Jan 30;24(3). 2606 10.3390/ijms24032606
- Cummins N, Tweedie A, Zuryn S, et al. Disease-associated tau impairs mitophagy by inhibiting Parkin translocation to mitochondria. EMBO J. 2019 Feb 1;38(3). 10.15252/embj.201899360
- Sík A, Passer BJ, Koonin EV, et al. Self-regulated cleavage of the mitochondrial intramembrane-cleaving protease PARL yields Pbeta, a nuclear-targeted peptide. J Biol Chem. 2004 Apr 9;279(15):15323–9.
- Jeyaraju DV, Xu L, Letellier MC, et al. Phosphorylation and cleavage of presenilin-associated rhomboid-like protein (PARL) promotes changes in mitochondrial morphology. Proc Natl Acad Sci U S A. 2006 Dec 5;103(49):18562–18567. 10.1073/pnas.0604983103
- Stringer BW, Day BW, D’Souza RCJ, et al. A reference collection of patient-derived cell line and xenograft models of proneural, classical and mesenchymal glioblastoma. Sci Rep. 2019 Mar 20;9(1):4902.10.1038/s41598-019-41277-z
- Ma Y, Zhang L, Huang X Genome modification by CRISPR/Cas9. FEBS J. 2014 Dec;281(23):5186–5193. 10.1111/febs.13110
- Lu G, Tan HWS, Schmauck-Medina T, et al. WIPI2 positively regulates mitophagy by promoting mitochondrial recruitment of VCP. Autophagy. 20222022/12 /02;18(12):2865–2879.
- Tan HWS, Lu G, Dong H, et al. A degradative to secretory autophagy switch mediates mitochondria clearance in the absence of the mATG8-conjugation machinery. Nature Communications. 20222022/06 /28;13(1):3720.
- Wang L, Cho YL, Tang Y, et al. PTEN-L is a novel protein phosphatase for ubiquitin dephosphorylation to inhibit PINK1-Parkin-mediated mitophagy. Cell Res. 2018 Aug;28(8):787–802.
- Zhang T, Xue L, Li L, et al. BNIP3 Protein Suppresses PINK1 Kinase Proteolytic Cleavage to Promote Mitophagy. J Biol Chem. 2016 Oct 7;291(41):21616–21629. 10.1074/jbc.M116.733410
- Chen ML, Shah V, Patnaik R, et al. Bioavailability and bioequivalence: an FDA regulatory overview. Pharm Res. 2001 Dec;18(12):1645–1650. 10.1023/A:1013319408893
- Brenner S The genetics of Caenorhabditis elegans. Genetics. 1974 May;77(1):71–94.10.1093/genetics/77.1.71
- Cao SQ, Wang HL, Palikaras K, et al. Chemotaxis assay for evaluation of memory-like behavior in wild-type and Alzheimer’s-disease-like C. elegans models. STAR Protoc. 2023 Apr 26;4(2):102250.
- Porta-de-la-Riva M, Fontrodona L, Villanueva A, et al. Basic Caenorhabditis elegans methods: synchronization and observation. J Vis Exp. 2012 Jun 10(64):e4019. 10.3791/4019
- Winston WM, Molodowitch C, Hunter CP Systemic RNAi in C. elegans requires the putative transmembrane protein SID-1. Science. 2002 Mar 29;295(5564):2456–2459.10.1126/science.1068836
- Calixto A, Chelur D, Topalidou I, et al. Enhanced neuronal RNAi in C. elegans using SID-1. Nat Methods. 2010 Jul;7(7):554–559.10.1038/nmeth.1463
- Liu F, Xiao Y, Ji XL, et al. The cAMP-PKA pathway-mediated fat mobilization is required for cold tolerance in C. elegans. Sci Rep. 2017 Apr 4;7(1):638.10.1038/s41598-017-00630-w
- Sutphin GL, Kaeberlein M Measuring Caenorhabditis elegans life span on solid media. J Vis Exp. 2009 May 12(27). 10.3791/1152-v
- Voglis G, Tavernarakis N A synaptic DEG/ENaC ion channel mediates learning in C. elegans by facilitating dopamine signalling. EMBO J. 2008 Dec 17;27(24):3288–3299.10.1038/emboj.2008.252
- Bargmann CI, Horvitz HR Chemosensory neurons with overlapping functions direct chemotaxis to multiple chemicals in C. elegans. Neuron. 1991 Nov;7(5):729–742.10.1016/0896-6273(91)90276-6
- Fang EF, Kassahun H, Croteau DL, et al. NAD(+) Replenishment Improves Lifespan and Healthspan in Ataxia Telangiectasia Models via Mitophagy and DNA Repair. Cell Metab. 2016 Oct 11;24(4):566–581.
- Ianevski A, Kulesskiy E, Krpina K, et al. Chemical, Physical and Biological Triggers of Evolutionary Conserved Bcl-xL-Mediated Apoptosis. Cancers (Basel). 2020 Jun 25;12(6). 1694 10.3390/cancers12061694