
ABSTRACT
Hepatocellular carcinoma (HCC) is a deadly malignancy characterized at the epigenetic level by global DNA hypomethylation and focal hypermethylation on the promoter of tumor suppressor genes. In most cases it develops on a background of liver steatohepatitis, fibrosis, and cirrhosis. Guadecitabine (SGI-110) is a second-generation hypomethylating agent, which inhibits DNA methyltransferases. Guadecitabine is formulated as a dinucleotide of decitabine and deoxyguanosine that is resistant to cytidine deaminase (CDA) degradation and results in prolonged in vivo exposure to decitabine following small volume subcutaneous administration of guadecitabine. Here we found that guadecitabine is an effective demethylating agent and is able to prevent HCC progression in pre-clinical models. In a xenograft HCC HepG2 model, guadecitabine impeded tumor growth and inhibited angiogenesis, while it could not prevent liver fibrosis and inflammation in a mouse model of steatohepatitis. Demethylating efficacy of guadecitabine on LINE-1 elements was found to be the highest 8 d post-infusion in blood samples of mice. Analysis of a panel of human HCC vs. normal tissue revealed a signature of hypermethylated tumor suppressor genes (CDKN1A, CDKN2A, DLEC1, E2F1, GSTP1, OPCML, E2F1, RASSF1, RUNX3, and SOCS1) as detected by methylation-specific PCR. A pronounced demethylating effect of guadecitabine was obtained also in the promoters of a subset of tumor suppressors genes (CDKN2A, DLEC1, and RUNX3) in HepG2 and Huh-7 HCC cells. Finally, we analyzed the role of macroH2A1, a variant of histone H2A, an oncogene upregulated in human cirrhosis/HCC that synergizes with DNA methylation in suppressing tumor suppressor genes, and it prevents the inhibition of cell growth triggered by decitabine in HCC cells. Guadecitabine, in contrast to decitabine, blocked growth in HCC cells overexpressing macroH2A1 histones and with high CDA levels, despite being unable to fully demethylate CDKN2A, RUNX3, and DLEC1 promoters altered by macroH2A1. Collectively, our findings in human and mice models reveal novel epigenetic anti-HCC effects of guadecitabine, which might be effective specifically in advanced states of the disease.
Introduction
Hepatocellular carcinoma (HCC) is the second leading cause of cancer-related death and the sixth most frequently diagnosed cancer worldwide. Prognosis is poor, as only 10 to 20% of patients with HCC are eligible for surgery; without it, patients are expected to survive less than 6 months.Citation1 HCC is often triggered, in an age-dependent manner, by viral infection, metabolic syndrome, and non-alcoholic fatty liver disease (NAFLD), along with the insurgence of cirrhosis.Citation2 There is no clear understanding of the epigenetic alterations in HCC or the potential role of DNA methylation markers as prognostic biomarkers. Nonetheless, global DNA hypomethylation, accompanied by focal hypermethylation, is generally found in HCC compared to healthy liver tissue.Citation3-5 Histone variants provide continuous regulation of nucleosome turnover across the entire lifespan of the organism in vertebrates, and they are a primary cellular strategy to regulate transcription and cellular metabolism.Citation6 Specifically, the histone H2A variant macroH2A1, comprising 2 alternatively spliced isoforms, macroH2A1.1 and macroH2A1.2, is a marker of human HCC.Citation7 MacroH2A1 isoforms have taken center stage in the plasticity of stem cell differentiation and in the pathogenesis of many cancers, providing an exciting, yet poorly understood, link to metabolism and nutrients.Citation8 Interestingly, macroH2A1 function and DNA methylation are functionally linked: in human fibroblasts, macroH2A1 deficiency combined with 5-aza-deoxycytidine (5-aza-dC, decitabine) treatment activates tumor-suppressor genes (TSG) and inhibits cell proliferation.Citation9 In the presence of HCC-associated DNA hypomethylation, high levels of macroH2A1 favor HCC progression through a distinct p38 MAPK/IL8-dependent mechanism that is accompanied by escape from senescence.Citation3 Hypomethylating agent decitabine and its derivatives inhibit DNA methyltransferases proteins and are mainly used for the treatment of myeloid malignancies, such as myelodisplastic syndrome (MDS) and acute myeloid leukemia (AML), although they also exert antitumor effects in vitro by inducing senescence.Citation10-14 Guadecitabine is a second-generation hypomethylating agent designed as a dinucleotide of decitabine and deoxyguanosine that is resistant to cytidine deaminase (CDA) Citation15 and results in prolonged in vivo exposure to decitabine following small volume subcutaneous administration of guadecitabine. Safety and clinical activity in MDS and AML have been shown in a phase 1 trial.Citation16,17 CDA rapidly metabolizes decitabine into inactive uridine counterparts, severely limiting exposure time and oral bioavailability. High expression of CDA is a distinctive feature of poor-prognosis leukemias.Citation18 In the liver, high CDA expression provides a “sanctuary” for cancer cells from decitabine treatment effects; this mechanism of resistance could be reversed in murine models, without increasing toxicity, by combining decitabine with an inhibitor of CDA,Citation19 a strategy now employed in ongoing clinical trials for myelodisplastic syndromes.Citation20,21 Preliminary evidence suggests that guadecitabine synergizes with other chemotherapeutics (sorafenib, oxaliplatin) in slowing down HCC cell growth in vitroCitation13,14 and a phase 2 clinical study evaluated guadecitabine single agent activity in patients with HCC after progression on sorafenib.Citation22 The aims of this study were to investigate the efficacy of guadecitabine in pre-clinical HCC models and to unravel its interplay with macroH2A1 oncogenes in regulating TSG promoter methylation and cancer growth. To this purpose we used xenograft and chemical/dietary mice models of liver diseases, macroH2A1-isoform specific transgenic HCC cell lines, and human liver tissues. We found that guadecitabine administration to decitabine resistant macroH2A1-overexpressing HCC cells and to HCC xenograft mouse model was able to hamper cancer growth, while it had not therapeutic effect on steatohepatitis.
Results
Guadecitabine (SGI-110) treatment inhibits HepG2 cell growth and vascularization in immunodeficient mice but does not affect pre-neoplastic stages in a dietary NASH model.
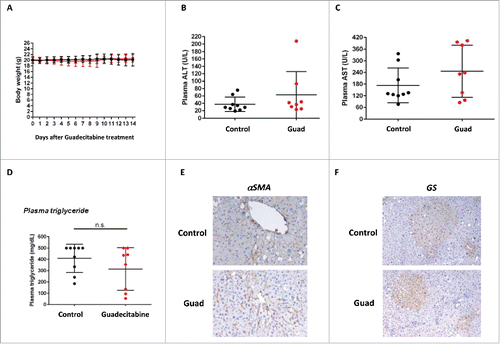
To investigate the in vivo antitumor efficacy of guadecitabine, xenograft studies were performed in athymic nude mice.Citation13 Subcutaneous HCC xenografts from HepG2 cells were established on the dorsal flank of balb/c immunodeficient mice, and treated with guadecitabine until tumor size in the control/untreated group reached 1400 mm3 (approximately 28 d post-inoculation). Treatment was given in a 3-day cycle, where guadecitabine (2 mg/kg) was given daily on Day 1–3. This mode of administration was chosen because in this mouse strain led to consistent LINE-1 demethylation in peripheral blood mononuclear cells, indicative of drug systemic efficacy, which was greater 9 d than 16 d post-treatment (). Xenografted mice were sacrificed and samples were collected when tumor sizes reached 1400 mm3 in the vehicle (PBS-injected) group, approximately 4 weeks post-inoculation. Organ sections of liver, kidney, heart, lung, spleen, and pancreas did not reveal major histopathological changes (data not shown). Tumor burden could be assessed only in 9 of the 14 mice of the guadecitabine group, whereas all 14 PBS-injected mice developed tumors. Tumor size was variable both within the PBS-injected and within the guadecitabine-injected groups (); no statistical difference in tumor size between the 2 experimental groups has been observed. However, a strikingly different weight range distribution was found, as 71% (10 out of 14) of the tumors of the PBS group compared to 33% (3 out of 9) of the tumors of the guadecitabine group were heavier than 0.2 g (). LINE-1 were found consistently demethylated in HepG2-derived tumors from guadecitabine treated animals compared to PBS-injected ones (Fig. S1A). Moreover, analysis of a DNA methylation array of 22 human tumor suppressor genes involved in HCC uncovered high hypermethylation levels of DAB2IP, DLEC1, GSTP1, RASSF1, RUNX3, and SOCS1 promoters in the HepG2-derived tumors from PBS-injected animals; these promoters were significantly demethylated upon guadecitabine treatment (Fig. S1B). Guadecitabine thus suppressed the growth of tumors, as detected at sacrifice, with therapeutic responsiveness detected at the level of both global and focal DNA methylation. Isolectin B4 fluorescent staining showed that the densities of the capillaries in the HepG2 xenografts of control PBS-injected animals were much higher compared to guadecitabine-injected animals (). Therefore, guadecitabine inhibits HCC growth and angiogenesis in a xenograft model. Non-alcoholic fatty liver disease (NAFLD), an accumulation of intra-hepatic triglycerides, is the most common cause of chronic liver disease in Western countries with up to one third of the population affected. NAFLD include a spectrum of disturbances that encompasses various degrees of liver damage ranging from simple steatosis to non-alcoholic steatohepatitis (NASH).Citation23 NASH is characterized by hepatocellular inflammation with fibrosis. Individuals with NASH are also at great risk of developing HCC. For this reason, here we used the STAM mouse, a model of NASH progression resembling the disease in humans.Citation24 On the second day after birth, male mice were subjected to a single subcutaneous injection of 200 μg streptozotocin. Four weeks after injection, mice were fed high fat diet (60% energy from lard) ad libitum until sacrifice at 10 weeks. Two experimental groups were considered, a control group (n = 9) and a group injected SC with guadecitabine (2 mg/kg, guadecitabine) (n = 8) at week 8 for 3 consecutive days. All mice were sacrificed at week 10. These time points were chosen because well characterized STAM mice develop NASH at week 8 and fibrosis, inflammation, and pre-neoplastic foci at week 10–12 (data not shown andCitation24,15). Guadecitabine administration led to ∼9% decrease in LINE-1 methylation levels in the livers of STAM mice as compared to age-matched mice fed a chow diet (controls); STAM mice, in turn, displayed LINE-1 methylation levels similar to those of controls (Fig. S2A). Surprisingly, despite unaltered LINE-1 methylation levels when comparing diets, the livers of STAM mice exhibited an increase in the maintenance DNA methyltransferase protein, DNMT1, compared to control mice (Fig. S2B). Upon administration of guadecitabine, DNMT1 protein levels returned to basal (Fig. S2B). Guadecitabine did not affect body weight () or the levels of circulating transaminases ALT and AST (), and did not induce changes in plasma triglycerides (). Immunopositivity for α-smooth muscle actin (αSMA), a marker of liver fibrosis, was observed in STAM mice, without tangible differences between control and guadecitabine-treated mice (). Moreover, glutamine synthetase positive foci were found in the livers of STAM mice. Glutamine synthetase foci represent putative preneoplastic and neoplastic lesions during initial stages of hepatocarcinogenesis: no differences were observed upon guadecitabine injections (). We conclude that although guadecitabine is not hepatotoxic and could be effective in reducing tumor mass, it might not be effective in alleviating inflammation and fibrosis in the experimental models used.
Figure 1. Relative changes in LINE-1 methylation levels in blood samples of balb/c mice treated with guadecitabine. Two months old female female Balb/c nude mice (BALB/cOlaHsd-Foxn1nu)balb/c Huh-7 tumor-bearing mice were injected s.c. with 2 mg/kg of guadecitabine and LINE-1 demethylation levels in whole blood [calculated as (methylation levels at day x – methylation levels at day 0)/(methylation levels day 0 *100)] were assessed at 9 and at 16 d post guadecitabine administration. Results for individual mice are shown. Six out of 8 mice (75%) show a distinct relative LINE-1 demethylation.
![Figure 1. Relative changes in LINE-1 methylation levels in blood samples of balb/c mice treated with guadecitabine. Two months old female female Balb/c nude mice (BALB/cOlaHsd-Foxn1nu)balb/c Huh-7 tumor-bearing mice were injected s.c. with 2 mg/kg of guadecitabine and LINE-1 demethylation levels in whole blood [calculated as (methylation levels at day x – methylation levels at day 0)/(methylation levels day 0 *100)] were assessed at 9 and at 16 d post guadecitabine administration. Results for individual mice are shown. Six out of 8 mice (75%) show a distinct relative LINE-1 demethylation.](/cms/asset/f4913848-a30e-47e7-b320-2a498b43fdc8/kepi_a_1214781_f0001_oc.gif)
Figure 2. Guadecitabine (Guad) prevents tumor growth in a HepG2 xenograft model. A. Pictures of explanted HepG2 tumors from PBS- and guadecitabine-injected immunocompromised mice, next to a ruler. B. Tumor growth in balb/c mice inoculated with HepG2 cells. The control group (n = 14) was injected with PBS while the experimental group (n = 14) was injected daily with 2 mg/kg guadecitabine from day 1 to 3 post-inoculation. Animals were sacrificed after 38 d, tumors were excised, and tumor, and volume/weight were assessed by means of a caliper and a precision scale, respectively. C. Tumors as in A-B were processed for H&E staining or for immunofluorescence with antibody against IsolectinB4 (IB4). For IF staining, nuclei were counterstained with DAPI.
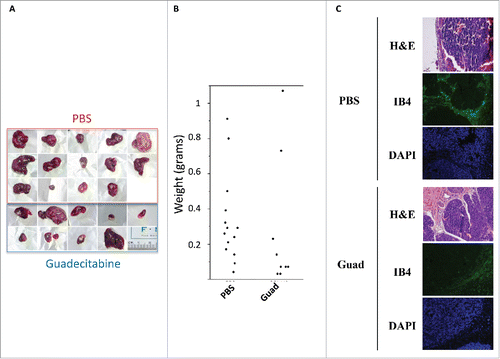
Figure 3. Guadecitabine (Guad) treatment does not influence liver fibrosis and preneoplastic foci formation in a NASH mouse model (STAM). The experimental group of mice was injected at week 8 with 2 mg/kg guadecitabine and sacrificed at week 10. A. Mice body weights were assessed daily after initial guadecitabine injection. B and C. ALT and AST transaminases levels (U/L) measured in mice sera upon sacrifice. D. Plasma triglycerides (mg/dL) measured in mice sera upon sacrifice. E. Immunohistochemical staining for αSMA in the livers of control vehicle-injected STAM mice (upper panel) or guadecitabine injected (lower panel). F. Immunohistochemical staining for glutamine synthetase (GS) in the livers of control vehicle-injected STAM mice (upper panel) or guadecitabine injected (lower panel). n = 9 for untreated STAM mice; n = 8 for guadecitabine-injected animals. Differences were not statistically significant (ns).
Human HCC methylation signature: effect of guadecitabine
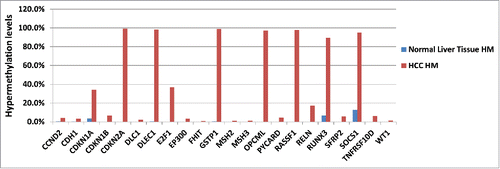
DNA methylation markers can be used as prognostic biomarkers for HCC. A recent analysis of tumor tissue from 304 patients with HCC treated with surgical resection generated a methylation-based prognostic signature that correlated with predictors of poor outcome and prognostic capacity of survival.Citation5 Patient derived xenografts (PDX), without in vitro manipulation, mirror patient histopathological and genetic profiles and are increasingly recognized to be predictive models for evaluating anti-cancer therapies. Here, we compared HCC PDX resistant to sorafenib (Nexavar®), a tyrosine kinase inhibitor approved for HCC therapy, to normal liver tissue from a healthy donor for possible differences in their DNA methylation signature, using a DNA methylation array of 22 human tumor suppressor genes involved in HCC (). Ten genes showed methylation differences between healthy liver tissue and malignant tissue: CDKN1A, CDKN2A, DLEC1, E2F1, GSTP1, OPCML, E2F1, RASSF1, RUNX3, and SOCS1 (). These genes, whose promoter is unmethylated in normal liver tissue and hypermethylated in HCC, might prove useful candidate markers to further investigate if different DNA methylation levels play a role in the sensitivity to chemotherapy in HCC.
Figure 4. HuPrime® PDX HCC Sorafenib resistant tumor DNA was compared with normal liver tissue from a healthy donor for differences in their DNA methylation signature shown as HM: hypermethylated levels for each marker gene. Liver Cancer EpiTect Methyl II PCR Arrays were used to measure CpG island methylation of a panel of 22 tumor suppressor genes involved in HCC development and progression (CCND2, CDH1, CDKN1A, CDKN1B, CDKN2A, DLC1, DLEC1, E2F1, EP300, FHIT, GSTP1, MSH2, MSH3, OPCML, SOCS1, E2F1, RASSF1, RELN, RUNX3, SFRP2, TNFRSF10D, WT1). Results for PDX are shown as the average of 8 individual sorafenib-resistant HCC models vs. methylation levels in healthy liver tissue.
Guadecitabine blocks the proliferation of decitabine-resistant HCC cells
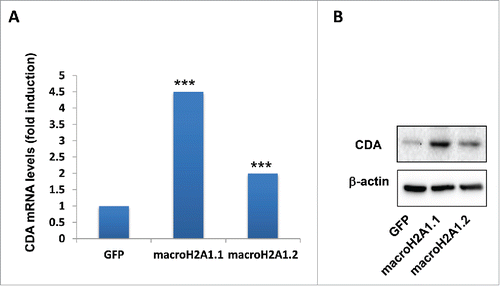
Histone variants macroH2A1.1 and macroH2A1.2 are strong immunohistochemical markers of human HCC, as they are found massively upregulated in malignant compared to cirrhotic and mildly steatotic HCC ().Citation3,7 Recently, we also demonstrated that these histone variants confer resistance to decitabine-induced senescence and block in cell growth, highjacking the senescence-associated secretory program in HCC cells.Citation3 We thus employed previously reported lentiviral-mediated stable HepG2 and Huh-7 cell lines transgenic for green fluorescent protein (GFP), macroH2A1.1-GFP, or macroH2A1.2-GFP ()Citation3 to study the functional differences between decitabine and guadecitabine. As expected, macroH2A1.1-GFP and macroH2A1.2-GFP transgene-bearing HepG2 and Huh-7 cells displayed a significant 2- to 3-fold increase in cell growth compared with control GFP-expressing cells after 12 μM decitabine treatment at 72 h, while there were no differences in the proliferation rate of untreated cells (). In contrast to decitabine, incubation with guadecitabine at lower doses of 1 μm and 5 μM blocked HepG2 and Huh-7 cell proliferation at 72 h to similar extent in all genotypes (). The only structural difference between guadecitabine and decitabine is the addition of a guanosine. Guadecitabine is resistant to inactivation by CDA,Citation15 which is highly expressed in liver and gut, and is a distinctive feature of poor-prognosis cancers.Citation18 We thus interrogated our previously generated RNA-Seq data of GFP, macroH2A1.1-GFP, or macroH2A1.2-GFP HepG2 cells for CDA expression, uncovering significantly higher mRNA expression of the enzyme in macro histone-overexpressing cells ().Citation3 We validated these changes in mRNA expression levels at the protein level by immunoblotting analysis, which showed consistent increase in CDA in decitabine-resistant macroH2A1-transgenic HepG2 cells compared to GFP control cells (). Our data suggest increased pharmacological responsiveness of HCC cells to guadecitabine compared to decitabine, despite CDA upregulation mediated by macroH2A1, which in turn is highly expressed in HCC.Citation3,7
Figure 5. Representative pictures of immunostainings for macroH2A1.1 (m1.1) and macroH2A1.2 (m1.2) in samples from patients diagnosed with hepatocellular carcinoma (HCC), cirrhosis, and steatosis. Bar: 100 μM. All nuclei of tumor cells were positive for either m1.1 or m1.2. Positivity in hepatocytes of cirrhosis and steatosis was significantly lower.
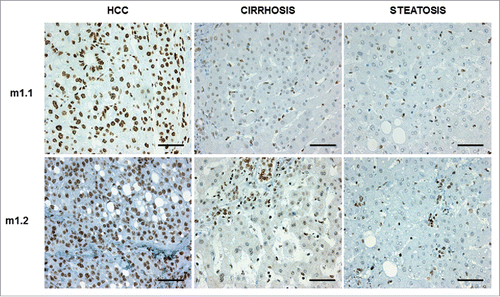
Figure 6. Representative images of HepG2 (A) and Huh-7 cells (B) transgenic for GFP, macroH2A1.1-GFP (macroH2A1.1) and macroH2A1.2-GFP (macroH2A1.2). Images were taken with respective channels for DAPI (blue) and GFP (green). C and D, MTT assay in HepG2 (C) and Huh-7 (D) cells after 72 h with decitabine (decit, 12 μM) or guadecitabine (SGI-110, 1 or 5 μM) incubation. Percentage growth is with untreated GFP control cells. All data were expressed as mean ± standard error of mean (SEM) of 4 independent experiments. *** P < 0.001 vs. untreated GFP-expressing cells. # P < 0.01 and ## P < 0.001 vs. decitabine-treated GFP-expressing cells.
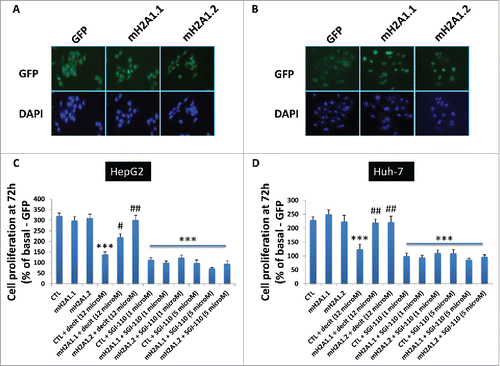
Figure 7. Cytidine deaminase (CDA) levels in HepG2 overexpressing GFP (control), macroH2A1.1 or macroH2A1.2 histone variants. A. CDA mRNA levels were averaged from 3 independent RNA-Seq experiments. B. CDA protein levels were analyzed by immunoblotting. β−actin was used as internal loading control. Representative images of 3 independent experiments are shown. *** P < 0.001 vs. GFP-expressing cells.
Guadecitabine-dependent promoter demethylation of tumor suppressor genes in HCC cells
Some of the epigenetic hallmarks of HCC are hypermethylation of TSGs and hypomethylation of unique genes and repetitive sequences, such as LINE-1, leading to global DNA hypomethylation. Our previous global analysis of DNA methylation using the Luminometric Methylation Assay (LUMA) showed similar genome-wide methylation levels in HepG2 and Huh-7 cells.Citation3 Basal-state analysis of 22 human tumor suppressor genes involved in HCC highlighted a similar trend of promoter methylation between the 2 cell lines for 16 tumor suppressor genes, while only 6 genes exhibited main differences in the levels of their hypermethylated promoters (Supplemental ). MacroH2A1-transgene overexpressing HepG2 and Huh-7 HCC cells were responsive to guadecitabine treatment (), and macroH2A1 and DNA methylation synergized in suppressing TSG genes expression.Citation9 Therefore, we sought to study DNA promoter hypermethylation from a subset of genes identified in human HCC tissues that were demethylated by guadecitabine (). To this purpose, we used sets of primers previously validated for their specificity by quantitative methylation-specific PCR () to amplify unmethylated or methylated CpG islands within the promoters of CDKN2A, DLEC1, and RUNX3, and we measured the unmethylated (U) vs. methylated (M) ratios in our HepG2 and Huh-7 cell lines upon prolonged exposure to guadecitabine (1 μM for 3 days). As expected, control GFP-overexpressing HepG2 and Huh-7 cells displayed demethylated levels of CDKN2A, DLEC1, and RUNX3 after treatment with guadecitabine (). At the basal state, TSG promoter methylation levels did not differ in macroH2A1.1- and macroH2A1.2-overexpressing HCC cells (). However, after treatment with guadecitabine, demethylation events on TSG promoters in macroH2A1 histones-overexpressing HCC cells were not occurring to the same extent or were discordant from control cells, with the exception of CDKN2A and DLEC1 in macroH2A1.2-overexpressing Huh-7 cells (). These data suggest that macroH2A1 histone upregulation (as observed in HCC tissue, ) directly or indirectly interfere with guadecitabine-induced TSG promoter demethylation.
Figure 8. Quantitative methylation-specific PCR of CpG islands of TSG (CDKN2A, DLEC1, and RUNX3) promoters in HCC cells. HepG2 cells (A) and Huh-7 cells (B) stably overexpressing GFP, macroH2A1.1-GFP, or macroH2A1.2-GFP were treated with 1 μM guadecitabine (SGI-110) for 72 h prior to DNA extraction, bisulfite conversion, and processing for qPCR. Primer sequences are shown in . Results are expressed as fold changes of controls (GFP) and as ratios between unmethylated (U) and methylated (M) DNA levels. All data were expressed as mean ± SEM of four independent experiments.* P < 0.05 vs. untreated GFP-expressing cells.
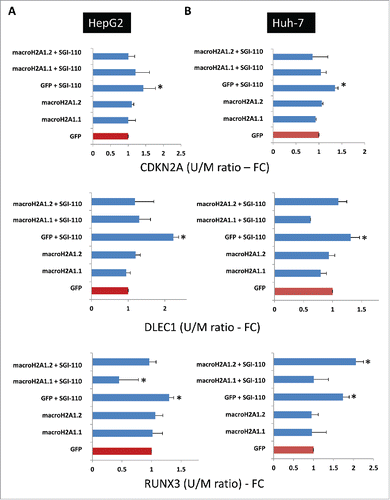
Table 1. qPCR primers for methylated/unmethylated gene promoters. Mf= methylated forward; Mr= methylated reverse; Uf: unmethylated forward; Ur: unmethylated reverse.
Discussion
The deregulation of epigenetic mechanisms, which maintain heritable gene expression changes and chromatin organization, is implicated in the development of HCC. Several independent genome-wide methylation profile studies demonstrated that HCC tumors display differential DNA methylation patterns compared to the adjacent normal liver tissues; these features have been proposed as tools for early diagnosis and prognosis of HCC.Citation3-5,26 Mechanistic studies involved chromatin remodeling complexes and microRNAs in HCC development, while the role of histone post-translational modifications, such as acetylation and methylation, is limited to correlative studies with clinical features of HCC. Most histones are synthesized at S phase for rapid deposition behind replication forks: the replacement of histones deposited during S phase by variants that can be deposited independently of replication provide one of the most fundamental level of chromatin differentiation.Citation27 Our laboratory demonstrated for the first time that the largest histone variant, namely macroH2A1, and its 2 splicing isoforms, macroH2A1.1 and macroH2A1.2, marks the progression of liver diseases from NAFLD to HCC.Citation3,6,7,28 While it has been shown that macroH2A1.1 acts as a tumor suppressor in many cancer types, the role of macroH2A1.2 is context-dependent.Citation8 Specifically, in HCC, macroH2A1 isoforms display similar and overlapping oncogenic roles, and there is a lack of linearity in macroH2A1 pre-mRNA splicing processes identified in malignant vs. adjacent normal parenchyma.Citation3,7,29 MacroH2A1 confers resistance to the anti-HCC activity of decitabine (5-Aza-2′-deoxycytidine), an epigenetic modifier that demethylates DNA and exerts tumor suppressor gene activation by remodeling.Citation10-12 MacroH2A1 overexpression in HCC cells leads to the hypersecretion of IL-8, which allows cancer cells to bypass the stall in proliferation/senescence induced by decitabine.Citation3 In the clinic, decitabine is successfully used for the treatment of myelodysplastic syndromes and for acute myeloid leukemia (AML), rather than for less proliferative diseases and for solid tumors, such as HCC, due to its short half-life in the blood: only 15 to 25 min, due to rapid inactivation by liver and gut CDA. For this reason, guadecitabine (SGI-110), a novel hypomethylating dinucleotide of decitabine and deoxyguanosine resistant to degradation by CDA, has been devised. In ongoing trials, guadecitabine was given on 5 consecutive days in a 28-day cycle, showing safety and tolerability in patients.Citation16,30 Recent studies showed that guadecitabine is effective in reducing tumor burdening by reprograming the epigenome as a single agent and in combination with other chemotherapeutic agents in immunocompromised mice xenografts and in cell models of lung cancer,Citation31 ovarian cancer,Citation17,26 and HCC.Citation13 In the current study, we confirmed the inhibitor effect of guadecitabine on HCC cancer growth in immunocompromised mice: smaller tumors developed in the presence of guadecitabine and were characterized by extensive demethylation of the promoters of tumor suppressor genes DAB2IP, DLEC1, GSTP1, RASSF1, RUNX3, and SOCS1. We also provided new evidence that the decrease in tumor burden might be due to a decrease in vascularization, as indicated by decreased isolectin B4 staining in tumor explants. Neo-angiogenesis is a key process in tumorigenesis; the methylated genomic targets of guadecitabine involved in angiogenesis and vascular sprouting remain to be elucidated. Furthermore, HepG2 and HuH-7 HCC cells overexpressing macroH2A1 isoforms that were refractory to decitabine treatment in vitro, displayed instead growth inhibition upon guadecitabine treatment, even at lower doses (1 and 5 μM) compared to decitabine (12 μM). We explain this discrepancy between decitabine and guadecitabine effects by the macroH2A1-dependent upregulation of CDA that emerged from RNA-Seq data and confirmed at the protein level in HepG2 cells overexpressing these histone variants.
HCC is a typical age-associated disease that develops over decades with strong risk factors coming from viral infections and alcohol consumption, and a common hallmark of a fibrotic/cirrhotic tissue background regardless of the etiology.Citation2,23,32 Here, we evaluated the efficacy of guadecitabine in blocking the progression of fibrosis in the STAM mouse, an established model of NASH, where fibrosis and inflammation are triggered by an initial injection of streptozocin in new born animals (which damage pancreatic β cells), followed by high fat diet feeding until 10 weeks of age. Despite using a dose (2 mg/kg) that correlates with the demethylation of LINE-1 elements in mice blood and liver (read-outs of its demethylating efficacy), guadecitabine treatment had no effect on liver fibrosis and proliferative foci formation in this NASH mouse model. We did not observe amelioration of transaminases levels, triglycerides, or in the appearance of histological markers of the disease, αSMA and glutamine synthetase, in the livers of control vehicle-injected and guadecitabine-injected mice. Liver fibrosis is orchestrated by the activation of hepatic stellate cells (HSC), responsible for extracellular matrix deposition.Citation33 In contrast to HCC, to date, genome-wide studies of changes in DNA methylation during HSC activation have not been reported and the prominent ones using human liver biopsies were to some extent conceptually and methodologically criticized;Citation34-37 therefore, changes in DNA methylation profiles might not play a role in hepatic fibrogenesis, a risk factor for HCC. However other studies showed that decitabine inhibit HSC activation.Citation37 Interestingly, the livers of STAM mice exhibited an increase in DNMT1, which has been consistently detected in human liver fibrosis or upon HCV infection,Citation38,39 compared to healthy mice livers. Administration of guadecitabine restored physiological DNMT1 levels; the precise role of DNMT1 in the transition from NASH to HCC and whether guadecitabine could influence the progression of liver disease remain unknown. An alternative explanation for the lack of guadecitabine effects on liver fibrosis in vivo is related to the high levels of CDA expression in this organ, which provides a hostile place for the stability of the drug and its effects on HSC and cancer cells. This mechanism of resistance could be reversed in mice, without toxicity, by combining decitabine with an inhibitor of CDA.Citation19 Combination of new DNA hypomethylating agents and CDA inhibitors is a new type of strategy now employed in ongoing clinical trials for several malignancies.Citation20 In fact, early studies using the CDA inhibitor tetrahydrouridine (THU) have shown proof of principle for combinatorial approach together with decitabine although its instability has prevented further development as a therapeutic.Citation40 Ongoing efforts to develop fluorinated derivatives of THU, which are resistant to degradation and can be co-administered with guadecitabine, will pave the way for more potent hypomethylation therapies against liver diseases and HCC. Regardless of clinical efficacy, DNA methylation markers can be used as prognostic biomarkers for HCC. Our methylation analysis showed differences in 10 genes between healthy liver tissue and malignant human HCC tissue: CDKN1A, CDKN2A, DLEC1, E2F1, GSTP1, OPCML, E2F1, RASSF1, RUNX3 and SOCS1. It would be interesting to follow up on these 10 differentially methylated markers in liver samples from a larger cohort of patients to screen for therapy responders/non-responders, and to study the correlation of these 9 markers with response/relapse. MacroH2A1 and DNA methylation synergize in suppressing TSG p16 expressionCitation9; therefore, we analyzed DNA promoter methylation levels from an arbitrary subset of 3 among these 10 genes identified in human HCC tissues that were demethylated by guadecitabine (CDKN2A, DLEC1, and RUNX3) in HCC cell lines, HepG2, and Huh-7 overexpressing histone variant macroH2A1 isoforms. At the basal level, control HepG2 and Huh-7 cell lines show similar HCC gene promoter methylation signature, with major differences observable only in 6 of the 22 genes analyzed (Supplemental Table 1), which can be possibly linked to the considerable differences in the genetic make up (p53 expression) and differentiation/proliferative behavior of these 2 cell lines.Citation41 In contrast to control cells, cells overexpressing macroH2A1 isoforms responded unevenly and, to a lesser extent, to guadecitabine in terms of TSG promoter demethylation, despite the fact that they were more responsive to guadecitabine compared to decitabine for inhibition of proliferation. This might be explained by i) wider guadecitabine-dependent DNA hypomethylation effects beyond the TSGs we analyzed, resulting in pleiotropic tumor suppression, overcoming macroH2A1 incorporation into chromatin, which might affect local demethylation on specific TSGs; and/or ii) intrinsic discrepancies in TSG methylation patterns between HCC cell lines and human biopsies. Chromatin co-immunoprecipitation studies using antibodies against macroH2A1 and DNA methylation marker 5-methylcytosine will help untangle these epigenetic interactions during HCC development. In summary, our data demonstrate that guadecitabine is effective in counteracting growth and vascularization in HCC xenografts and in HCC cells refractory to decitabine, while its efficacy as a single agent or in combination in pre-clinical models of fibrosis/cirrhosis requires further investigation. Effective hypomethylating agents will hopefully provide HCC patients with more potent therapeutic strategies in the future.
Materials and methods
Xenograft models
Female athymic nude mice (strain Hsd: Athymic Nude-Foxn1nu) were purchased from Harlan Laboratories (Horst, Netherlands) and allowed to acclimatize for a week at the Animal Facility of Tallinn University of Technology. The study was conducted in accordance with European Directive 2010/63/EU and National Animal Welfare legislation; the experimental protocol was approved by the ethical committee of Estonian Ministry of Agriculture (now Ministry of Rural Affairs). Animals were housed under 12 h light/dark cycles in a humidity and temperature-controlled room (temperature 22 ± 1°C and humidity 50 ± 10%). Mice were housed at 6–8 mice per cage with ad libitum access to clean water and food pellets (R35, Lactamin, Sweden) and their health was monitored daily. Six week-old mice were implanted subcutaneously (s.c.) with human hepatocellular carcinoma HepG2 cells stably expressing GFP.Citation3 For this, stable HepG2 cells were cultured to 70–80% confluency, harvested and suspended in complete media mixed 1:1 with Matrigel Matrix (Cat# 356234, Becton Dickinson, Bedford, MA). The suspension containing 7.5 × 106 cells (150 μl) was injected s.c. into the right flank of each mouse. Ten days later, mice were randomly assigned into the test group receiving guadecitabine treatment (n = 14), and the control group receiving vehicle (n = 14). For this, guadecitabine (provided by Astex Pharmaceuticals) reconstituted in its diluent buffer (containing 65% Propylene Glycol, 25% Glycerin, 10% Dehydrated Ethanol, 200 proof) was freshly diluted 100× in PBS and injected s.c. into the neck skin fold at 2 mg/kg and at a volume 10 ml/kg body weight on 5 consecutive days. About 15% of the guadecitabine-treated animals exhibited signs of weakness and 10% weight loss. Tumor sizes were measured 2–3 times per week with a digital caliper and the tumor volume was calculated using the ellipsoid formula: length × width 2 × 0.5. Mice were euthanized 5 weeks after xenograft implantation, and tumors were dissected, weighed, and processed for further analysis. Unpaired Student's t-test was performed for data analysis and P < 0.05 was considered significant.
Non-alcoholic steatohepatitis mouse model
The non-alcohonlic steatohepaptitis (NASH) model, a STAM mouse, was generated as previously describedCitation24 and outsourced at SMC Laboratories, Tokyo, Japan. Briefly, C57BL/6J mice were purchased from Charles River (Kanagawa, Japan) at 15 d post pregnancy. On the second day after birth, male mice were subjected to a single subcutaneous injection of 200 μg streptozotocin (Sigma, MO, USA). Four weeks after injection, mice were fed a high fat diet (HFD32, CLEA JAPAN, Tokyo, Japan) ad libitum until sacrifice at 12 weeks. Two experimental groups were considered, a control group (n = 9) and a group injected s.c. with guadecitabine (2 mg/kg, guadecitabine, Astex Pharmaceuticals) (n = 9) at week 8 for 3 consecutive days. Mice were sacrificed at week 10. Blood glucose and serum insulin levels were measured by a blood glucose meter (Glutest Ace, Sanwa Chemical, Nagoya, Japan) and Morinaga Ultra Sensitive Mouse/Rat Insulin ELISA Kit (Morinaga Institute of Biological Science, Yokohama, Japan), respectively. At 8 weeks, animal experiments were conducted according to protocols approved by the Animal Research Committee at Research Institute, National Center for Global Health and Medicine. Mice were maintained according to National Institutes of Health guidelines for care and use of laboratory animals.
LINE-1 methylation assay
Genomic DNAs were extracted from whole blood of 8 balb/c mice 8 d post s.c. injection with 2 mg/kg guadecitabine, using the Illustra blood genomic Prep Mini Spin Kit (GE Healthcare, UK) according to the manufacturer's protocol and stored at −20°C. DNA extracts were prepared from human HCC cell lines, human tumor and normal liver tissues, and from mice livers using the DNA Mini Kit (Qiagen). DNA concentrations were measured by NanoDrop analysis before the direct bisulfite conversion procedure using the EpiTect Fast DNA Bisulfite Kit (Qiagen), followed by a Mouse or a Human LINE-1 PCR reaction (EpigenDX). Methylation levels were analyzed on a PyroMarkQ24 instrument including methylation control DNAs to validate each sample set. The degree of methylation was expressed as percentage of methylated cytosines over the sum of methylated and unmethylated cytosines.
Tumor samples and methylation arrays
For human tumors, DNA was obtained from HuPrime® Patient-Derived Xenograft (PDX) Sorafenib-resistant HCC tissues (CrownBio International R&D Center, Taicang, P.R. China). DNA was also extracted from normal liver tissue (Donor HH180 lot 99; BD Bioscience, UK). For mice tumors, DNA was extracted from tumors of guadecitabine-injected and PBS-injected mice using the DNA Mini Kit (Qiagen). A DNA methylation signature for 22 genes was determined using the human EpiTect® DNA Methylation PCR Array platform for liver cancer (Qiagen) on a ViiA-7 RT-PCR instrument (Applied Biosciences).
Immunohistochemistry on steatotic, cirrhotic, and HCC human biopsies
Liver specimens were obtained from patients undergoing clinically indicated hepatic biopsies, with appropriate institutional (University of Palermo) ethical approval and patient consent and in accordance with the Code of Ethics of the World Medical Association (Declaration of Helsinki).Citation3,7 Liver sections in paraffin-embedded (4-μm thick, 10 per condition) were stained with hematoxylin and eosin (H&E) and Masson's trichrome for histologic evaluation, as described previously.Citation3,7 Diagnostic classification of NAFLD and fibrosis/cirrhosis was performed using a semiquantitative scoring system that grouped histologic features into broad categories (steatosis, hepatocellular injury, portal inflammation, fibrosis, and miscellaneous features).
Histology, immunohistochemistry, and immunofluorescence in mice
Tumor samples from xenograft models were formalin-fixed, paraffin-embedded and processed for H&E staining, using standard procedures. Isolectin IB4 protein was detected using Isolectin GS-IB4 Alexa fluor 488 conjugate antibody (Thermofisher Scientific, catalog number I21411) and incubated overnight at 4°C. Samples were then washed with PBS and nuclei were stained with DAPI for 5 min. Samples were finally mounted and conserved at −20°C before images were acquired using an optical microscope (Leica). Liver samples from the STAM model were fixed in 4% buffered paraformaldehyde, embedded in paraffin, stained with H&E or Masson's trichrome, and examined by light microscopy, as previously described.Citation24,25 Alternatively, samples were processed for immunohistochemical staining with antibodies against αSMA or glutamine synthetase.Citation24,25
Constructs, cell cultures, and proliferation assay
Stable expression of macroH2A1 isoforms tagged with GFP in HepG2 and Huh-7 cells was achieved by lentiviral transduction. cDNAs encoding macroH2A1 isoforms were placed under the control of a strong constitutive EF1α-promoter in the HIV1-based self-inactivating lentiviral vector. The vectors were modified to allow expression of macroH2A isoforms as N-terminal fusions with a monomeric fluorescent protein mTagGFP, and via the ECMV internal ribosomal entry site element of the puromycin resistance gene. All constructs were verified by sequencing. Viral preparations were produced by transient transfection in HEK293T cells, and concentrated 100-fold by low-speed centrifugation (6000 g for 16 h). HepG2 and Huh-7 cells were transduced with virus at MOI>10 for 24 h in the presence of 4 μg/mL Polybrene. Stably transduced cell populations were established and used in subsequent experiments without clonal selection. To generate appropriate controls, cells were also stably transduced with the empty vector (expressing only mTagGFP) and maintained in culture conditions similar to macroH2A1-expressing cells. For all experiments, cells were treated for 72 h with 5 or 10 μM of guadecitabine (SGI-110, Astex Pharmaceuticals). Cell proliferation was assessed by MTT assay, as previously described.Citation42,43
Methylation-specific PCR
DNA extracted from HepG2 and Huh-7 cells expressing GFP, macroH2A1.1-GFP or macroH2A1.2-GFP, was subjected to bisulphite treatment and DNA purification using the Epitect Bisulphite kit (Qiagen Sci, MD, USA) according to manufacturer instructions. Bisulfite-modified DNA was used as template for fluorescence-based real-time PCR using a relative quantification method.Citation44 Primers were designed to specifically amplify the bisulphite-modified region of the genes of interest containing the putative methylated CpGs. Primer sequences for the 5 genes whose promoters were examined are provided in . Amplification reactions were carried out in triplicate in a volume of 20 μL containing 50 ng bisulphite-modified DNA, 600 nM forward and reverse primers, and SYBR green mix (Qiagen Sci, MD, USA). PCR conditions were as follows: one step at 95°C for 3 min, 40 cycles at 95°C for 15 s, and 60°C to 62°C for 1 min. PCR reactions were performed in 96-well plates on a ABI PRISM 7700 Sequence detection system (Applied Biosystems, Carlsbad, CA) and were analyzed by SDS 2.1.1 software (Applied Biosystems, Foster City, CA).
RNA-Seq
For RNA-Seq, total RNA was extracted from macroH2A1.1-GFP, macroH2A1.2-GFP, and GFP transgenic HepG2 cell lines with TRIzol Reagent (Invitrogen). Indexed libraries were prepared from 2 mg/ea purified RNA with the TruSeq Total Stranded RNA Sample Prep Kit (Illumina), according to the manufacturer's instructions. Libraries were quantified using the Agilent 2100 Bioanalyzer (Agilent Technologies) and pooled so that each index-tagged sample was present in equimolar amounts; the final concentration of the pooled samples was 2 nmol/L. Pooled samples were then subjected to cluster generation and sequencing using an Illumina HiSeq 2500 System (Illumina, Genomix4Life, Baronissi, Salerno, Italy) in a 2 × 100 paired-end format at a final concentration of 8 pmol/L.
Western blot
Protein extracts from macroH2A1.1-GFP, macroH2A1.2-GFP, and GFP transgenic HepG2 and Huh-7 cells were isolated and processed for immunoblotting as described previously.Citation3,45 Primary antibody against CDA and DNA methyltransferases (DNMT1, DNMT3a, and DNMT3b) were obtained from Activ Motif and Cell Signaling, respectively. Antibody against β-actin (Santa Cruz Biotechnology) was used as total protein loading control for normalization.
Statistical analyses
Results were expressed as mean ± standard error of the mean (SEM). Comparisons between means were made by appropriate Student t tests. Differences of proportions were assessed by one-tailed χ2 tests. Differences were considered as significant when P < 0.05.
Disclosure of potential conflicts of interest
S. Jueliger, J. Lyons M. Azab and P. Taverna are employees of Astex Pharmaceuticals, Inc. The other authors disclose no potential conflicts of interest.
Author contributions
SJ and MV conceived and designed the study. SJ, SC, IP, PP, MS, OLR, MP and FR acquired data. SJ, PT, IP, PP, MS, VP, FV, FC, MA, JL and MV interpreted data. SJ and MV wrote the manuscript. SJ and MV supervised the study. All authors read and approved the manuscript.
KEPI_A_1214781_s02.zip
Download Zip (159.1 KB)Funding
PV and MV are supported by Italian Ministry of Health, Bando GR-2010-2311017. MV is supported by a My First Associazione Italiana Ricerca sul Cancro (AIRC) Grant-AIRC Grant No.13419, by UCL and by grants No.LQ1605 from the National Program of Sustainability II (MEYS CR) and FNUSA-ICRC No. CZ.1.05/1.1.00/02.0123 (OP VaVpI). PP is supported by EU-FP7 Grant No. 621364 (TUTIC-Green). This study was funded in part by Astex Pharmaceuticals. We are grateful to SMC Laboratories for assistance with the NASH mouse model, and to Dr. Concetta Panebianco for technical help.
Related Research Data
References
- Gores GJ. Decade in review-hepatocellular carcinoma: HCC-subtypes, stratification and sorafenib. Nat Rev Gastroenterol Hepatol 2014; 11(11):645-7; PMID:25245016; http://dx.doi.org/10.1038/nrgastro.2014.157
- Sheedfar F, Di Biase S, Koonen D, Vinciguerra M. Liver diseases and aging: friends or foes? Aging Cell 2013; 12(6):950-4; PMID:23815295; http://dx.doi.org/10.1111/acel.12128
- Borghesan M, Fusilli, C, Rappa, F, Panebianco, C, Rizzo, G, Oben, JA, Mazzoccoli, G, Faulkes, C, Pata, I, Agodi, A, et al. DNA Hypomethylation and Histone Variant macroH2A1 synergistically attenuate chemotherapy-induced senescence to promote hepatocellular carcinoma progression. Cancer Res 2016; 76(3):594-606; PMID:26772755; http://dx.doi.org/10.1158/0008-5472.CAN-15-1336
- Revill K, Wang T, Lachenmayer A, Kojima K, Harrington A, Li J, Hoshida Y, Llovet JM, Powers S. Genome-wide methylation analysis and epigenetic unmasking identify tumor suppressor genes in hepatocellular carcinoma. Gastroenterol 2013; 145(6):1424-35 e1-25; PMID:24012984; http://dx.doi.org/10.1053/j.gastro.2013.08.055
- Villanueva A, Portela A, Sayols S, Battiston C, Hoshida Y, Mendez-Gonzalez J, Imbeaud S, Letouzé E, Hernandez-Gea V, Cornella H, et al. DNA methylation-based prognosis and epidrivers in hepatocellular carcinoma. Hepatol 2015; 61(6):1945-56; PMID:25645722; http://dx.doi.org/10.1002/hep.27732
- Borghesan M, Mazzoccoli G, Sheedfar F, Oben J, Pazienza V, Vinciguerra M. Histone variants and lipid metabolism. Biochem Soc Trans 2014; 42(5):1409-13; PMID:25233423; http://dx.doi.org/10.1042/BST20140119
- Rappa F, Greco A, Podrini C, Cappello F, Foti M, Bourgoin L, Peyrou M, Marino A, Scibetta N, Williams R, et al. Immunopositivity for histone macroH2A1 isoforms marks steatosis-associated hepatocellular carcinoma. PLoS One 2013; 8(1):e54458; PMID:23372727; http://dx.doi.org/10.1371/journal.pone.0054458
- Cantarino N, Douet J, Buschbeck M. MacroH2A–an epigenetic regulator of cancer. Cancer Lett 2013; 336(2):247-52; PMID:23531411; http://dx.doi.org/10.1016/j.canlet.2013.03.022
- Barzily-Rokni M, Friedman N, Ron-Bigger S, Isaac S, Michlin D, Eden A. Synergism between DNA methylation and macroH2A1 occupancy in epigenetic silencing of the tumor suppressor gene p16(CDKN2A). Nucleic Acids Res 2011; 39(4):1326-35; PMID:21030442; http://dx.doi.org/10.1093/nar/gkq994
- Venturelli S, Berger A, Weiland T, Essmann F, Waibel M, Nuebling T, Häcker S, Schenk M, Schulze-Osthoff K, Salih HR, et al. Differential induction of apoptosis and senescence by the DNA methyltransferase inhibitors 5-azacytidine and 5-aza-2′-deoxycytidine in solid tumor cells. Mol Cancer Ther 2013; 12(10):2226-36; PMID:23924947; http://dx.doi.org/10.1158/1535-7163.MCT-13-0137
- Venturelli S, Berger A, Weiland T, Zimmermann M, Hacker S, Peter C, Wesselborg S, Königsrainer A, Weiss TS, Gregor M, et al. Dual antitumour effect of 5-azacytidine by inducing a breakdown of resistance-mediating factors and epigenetic modulation. Gut 2011; 60(2):156-65; PMID:21106551; http://dx.doi.org/10.1136/gut.2010.208041
- Venturelli S, Armeanu S, Pathil A, Hsieh CJ, Weiss TS, Vonthein R, Wehrmann M, Gregor M, Lauer UM, Bitzer M. Epigenetic combination therapy as a tumor-selective treatment approach for hepatocellular carcinoma. Cancer 2007; 109(10):2132-41; PMID:17407132; http://dx.doi.org/10.1002/cncr.22652
- Kuang Y, El-Khoueiry A, Taverna P, Ljungman M, Neamati N. Guadecitabine (SGI-110) priming sensitizes hepatocellular carcinoma cells to oxaliplatin. Mol Oncol 2015; 9(9):1799-814; PMID:26160429; http://dx.doi.org/10.1016/j.molonc.2015.06.002
- Jueliger S, Lyons J, Azab M, Taverna P. SGI-110, a novel second generation DNA hypomethylating agent, enhances sorafenib activity and alters the methylation signature of HCC cell lines. EORTC-AACR- NCI Symposium on Molecular Targets and Cancer Therapeutics Dublin, Ireland 2012.
- Coral S, Parisi G, Nicolay HJ, Colizzi F, Danielli R, Fratta E, Covre A, Taverna P, Sigalotti L, Maio M. Immunomodulatory activity of SGI-110, a 5-aza-2′-deoxycytidine-containing demethylating dinucleotide. Cancer Immunol Immunother 2013; 62(3):605-14; PMID:23138873; http://dx.doi.org/10.1007/s00262-012-1365-7
- Issa JP, Roboz G, Rizzieri D, Jabbour E, Stock W, O'Connell C, Yee K, Tibes R, Griffiths EA, Walsh K, et al. Safety and tolerability of guadecitabine (SGI-110) in patients with myelodysplastic syndrome and acute myeloid leukaemia: a multicentre, randomised, dose-escalation phase 1 study. Lancet Oncol 2015; 16(9):1099-110; PMID:26296954; http://dx.doi.org/10.1016/S1470-2045(15)00038-8
- Srivastava P, Paluch BE, Matsuzaki J, James SR, Collamat-Lai G, Karbach J, Nemeth MJ, Taverna P, Karpf AR, Griffiths EA. Immunomodulatory action of SGI-110, a hypomethylating agent, in acute myeloid leukemia cells and xenografts. Leuk Res 2014; 38(11):1332-41; PMID:25260825; http://dx.doi.org/10.1016/j.leukres.2014.09.001
- McCarthy H, Wierda WG, Barron LL, Cromwell CC, Wang J, Coombes KR, Rangel R, Elenitoba-Johnson KS, Keating MJ, Abruzzo LV. High expression of activation-induced cytidine deaminase (AID) and splice variants is a distinctive feature of poor-prognosis chronic lymphocytic leukemia. Blood 2003; 101(12):4903-8; PMID:12586616; http://dx.doi.org/10.1182/blood-2002-09-2906
- Ebrahem Q, Mahfouz RZ, Ng KP, Saunthararajah Y. High cytidine deaminase expression in the liver provides sanctuary for cancer cells from decitabine treatment effects. Oncotarget 2012; 3(10):1137-45; PMID:23087155; http://dx.doi.org/10.18632/oncotarget.597
- Maio M, Covre A, Fratta E, Di Giacomo AM, Taverna P, Natali PG, Coral S, Sigalotti L. Molecular pathways: At the crossroads of cancer epigenetics and immunotherapy. Clin Cancer Res 2015; 21(18):4040-7; PMID:26374074; http://dx.doi.org/10.1158/1078-0432.CCR-14-2914
- Savona MR, Malcovati L, Komrokji R, Tiu RV, Mughal TI, Orazi A, Kiladjian JJ, Padron E, Solary E, Tibes R, et al. An international consortium proposal of uniform response criteria for myelodysplastic/myeloproliferative neoplasms (MDS/MPN) in adults. Blood 2015; 125(12):1857-65; PMID:25624319; http://dx.doi.org/10.1182/blood-2014-10-607341
- El-Khoueiry AM, Bekali-Saab M, Kim T, Denlinger R, Goel CS, Gupta R, Jueliger S, Dua S, Oganesian R, Keer A, editor Single Agent Activity Of The Second-Generation Hypomethylating Agent, SGI-110, In Patients With Hepatocellular Carcinoma (HCC) After Progression On Sorafenib. 9th ILCA Annual Conference; 2015
- Podrini C, Borghesan M, Greco A, Pazienza V, Mazzoccoli G, Vinciguerra M. Redox homeostasis and epigenetics in non-alcoholic fatty liver disease (NAFLD). Curr Pharm Des 2013; 19(15):2737-46; PMID:23092327; http://dx.doi.org/10.2174/1381612811319150009.
- Saito K, Uebanso T, Maekawa K, Ishikawa M, Taguchi R, Nammo T, Nishimaki-Mogami T, Udagawa H, Fujii M, Shibazaki Y, et al. Characterization of hepatic lipid profiles in a mouse model with nonalcoholic steatohepatitis and subsequent fibrosis. Sci Rep 2015; 5:12466.
- Takakura K, Koido S, Fujii M, Hashiguchi T, Shibazaki Y, Yoneyama H, et al. Characterization of non-alcoholic steatohepatitis-derived hepatocellular carcinoma as a human stratification model in mice. Anticancer Res 2014; 34(9):4849-55; PMID:25202066
- Fang F, Munck J, Tang J, Taverna P, Wang Y, Miller DF, Pilrose J, Choy G, Azab M, Pawelczak KS, et al. The novel, small-molecule DNA methylation inhibitor SGI-110 as an ovarian cancer chemosensitizer. Clin Cancer Res 2014; 20(24):6504-16; PMID:25316809; http://dx.doi.org/10.1158/1078-0432.CCR-14-1553
- Henikoff S, Smith MM. Histone variants and epigenetics. Cold Spring Harb Perspect Biol 2015; 7(1):a019364; PMID:25561719; http://dx.doi.org/10.1101/cshperspect.a019364
- Podrini C, Koffas A, Chokshi S, Vinciguerra M, Lelliott CJ, White JK, Adissu HA, Williams R, Greco A. MacroH2A1 isoforms are associated with epigenetic markers for activation of lipogenic genes in fat-induced steatosis. FASEB J 2015; 29(5):1676-87; PMID:25526730; http://dx.doi.org/10.1096/fj.14-262717
- Novikov L, Park JW, Chen H, Klerman H, Jalloh AS, Gamble MJ. QKI-mediated alternative splicing of the histone variant MacroH2A1 regulates cancer cell proliferation. Mol Cell Biol 2011; 31(20):4244-55; PMID:21844227; http://dx.doi.org/10.1128/MCB.05244-11
- Srivastava P, Paluch BE, Matsuzaki J, James SR, Collamat-Lai G, Taverna P, Karpf AR, Griffiths EA. Immunomodulatory action of the DNA methyltransferase inhibitor SGI-110 in epithelial ovarian cancer cells and xenografts. Epigenetics 2015; 10(3):237-46; PMID:25793777; http://dx.doi.org/10.1080/15592294.2015.1017198
- Tellez CS, Grimes MJ, Picchi MA, Liu Y, March TH, Reed MD, Oganesian A, Taverna P, Belinsky SA. SGI-110 and entinostat therapy reduces lung tumor burden and reprograms the epigenome. Int J Cancer 2014; 135(9):2223-31; PMID:24668305; http://dx.doi.org/10.1002/ijc.28865
- Longo VD, Antebi A, Bartke A, Barzilai N, Brown-Borg HM, Caruso C, Curiel TJ, de Cabo R, Franceschi C, Gems D, et al. Interventions to slow aging in humans: Are we ready? Aging Cell 2015; 14(4):497-510; PMID:25902704; http://dx.doi.org/10.1111/acel.12338
- McKee C, Sigala B, Soeda J, Mouralidarane A, Morgan M, Mazzoccoli G, Rappa F, Cappello F, Cabibi D, Pazienza V, et al. Amphiregulin activates human hepatic stellate cells and is upregulated in non alcoholic steatohepatitis. Sci Rep 2015; 5:8812; PMID:25744849; http://dx.doi.org/10.1038/srep08812
- Moylan CA, Pang H, Dellinger A, Suzuki A, Garrett ME, Guy CD, Murphy SK, Ashley-Koch AE, Choi SS, Michelotti GA, et al. Hepatic gene expression profiles differentiate presymptomatic patients with mild vs. severe nonalcoholic fatty liver disease. Hepatol 2014; 59(2):471-82; PMID:23913408; http://dx.doi.org/10.1002/hep.26661
- Murphy SK, Yang H, Moylan CA, Pang H, Dellinger A, Abdelmalek MF, Garrett ME, Ashley-Koch A, Suzuki A, Tillmann HL, et al. Relationship between methylome and transcriptome in patients with nonalcoholic fatty liver disease. Gastroenterol 2013; 145(5):1076-87; PMID:23916847; http://dx.doi.org/10.1053/j.gastro.2013.07.047
- Vinciguerra M. Normalization of a NAFLD gene expression signature. Hepatol 2014; 60(4):1445; PMID:24493162; http://dx.doi.org/10.1002/hep.27042
- Mann DA. Epigenetics in liver disease. Hepatol 2014; 60(4):1418-25; PMID:24633972; http://dx.doi.org/10.1002/hep.27131
- Benegiamo G, Vinciguerra M, Mazzoccoli G, Piepoli A, Andriulli A, Pazienza V. DNA methyltransferases 1 and 3b expression in Huh-7 cells expressing HCV core protein of different genotypes. Dig Dis Sci 2012; 57(6):1598-603; PMID:22526584; http://dx.doi.org/10.1007/s10620-012-2160-1
- Bian EB, Huang C, Ma TT, Tao H, Zhang H, Cheng C, Lv XW, Li J. DNMT1-mediated PTEN hypermethylation confers hepatic stellate cell activation and liver fibrogenesis in rats. Toxicol Appl Pharmacol 2012; 264(1):13-22; PMID:22841775; http://dx.doi.org/10.1016/j.taap.2012.06.022
- Lavelle D, Vaitkus K, Ling Y, Ruiz MA, Mahfouz R, Ng KP, Negrotto S, Smith N, Terse P, Engelke KJ, et al. Effects of tetrahydrouridine on pharmacokinetics and pharmacodynamics of oral decitabine. Blood 2012; 119(5):1240-7; PMID:22160381; http://dx.doi.org/10.1182/blood-2011-08-371690
- Cervello M, Bachvarov D, Lampiasi N, Cusimano A, Azzolina A, McCubrey JA, Montalto G. Molecular mechanisms of sorafenib action in liver cancer cells. Cell Cycle 2012; 11(15):2843-55; PMID:22801548; http://dx.doi.org/10.4161/cc.21193
- D'Aronzo M, Vinciguerra M, Mazza T, Panebianco C, Saracino C, Pereira SP, Graziano P, Pazienza V. Fasting cycles potentiate the efficacy of gemcitabine treatment in in vitro and in vivo pancreatic cancer models. Oncotarget 2015; 6(21):18545-57; PMID:26176887; http://dx.doi.org/10.18632/oncotarget.4186
- Pazienza V, Borghesan M, Mazza T, Sheedfar F, Panebianco C, Williams R, Mazzoccoli G, Andriulli A, Nakanishi T, Vinciguerra M. SIRT1-metabolite binding histone macroH2A1.1 protects hepatocytes against lipid accumulation. Aging (Albany NY) 2014; 6(1):35-47; PMID:24473773; http://dx.doi.org/10.18632/aging.100632
- Ripoli M, Barbano R, Balsamo T, Piccoli C, Brunetti V, Coco M, Mazzoccoli G, Vinciguerra M, Pazienza V. Hypermethylated levels of E-cadherin promoter in Huh-7 cells expressing the HCV core protein. Virus Res 2011; 160(1–2):74-81; PMID:21640770; http://dx.doi.org/10.1016/j.virusres.2011.05.014
- Planavila A, Dominguez E, Navarro M, Vinciguerra M, Iglesias R, Giralt M, Lope-Piedrafita S, Ruberte J, Villarroya F. Dilated cardiomyopathy and mitochondrial dysfunction in Sirt1-deficient mice: a role for Sirt1-Mef2 in adult heart. J Mol Cell Cardiol 2012; 53(4):521-31; PMID:22986367; http://dx.doi.org/10.1016/j.yjmcc.2012.07.019