
ABSTRACT
Histone acetylation is regulated by histone acetyltransferases (HATs) and histone deacetylases (HDACs). It is associated with gene transcription and expression. 4-Phenylbutyric acid (4-PBA), an HDAC inhibitor (HDACi), can inhibit cancer cell proliferation by increasing the level of histone acetylation. However, 4-PBA did not show any efficacy in clinical trials. In this study, we found that 4-PBA induced epithelial–mesenchymal transition (EMT) in gastric cancer cell lines MGC-803 and BGC-823 with ectopic E-cadherin expression. Based on the expression profile microarray, IL-8 was the most significantly up-regulated gene by 4-PBA, and was selected for further investigation. Knockdown of IL-8 partially prevented 4-PBA-induced-EMT by blocking the activation of the downstream Gab2-ERK pathway. Furthermore, CHIP assay confirmed that acetyl-H3 directly combined with the promoter region of IL-8 to promote its transcription. Therefore, the results of this study demonstrated that 4-PBA-mediated inhibition of HDAC activity could induce EMT in gastric cancer cells via acetyl-histone-mediated IL-8 upregulation, and the downstream Gab2/ERK activation. These data indicated the possible reason for the failure of 4-PBA in clinical trials.
Introduction
Histone acetylation is one of the most important epigenetic modifications related to DNA replication and transcription. When acetylation modification is catalysed by histone acetyltransferase (HAT) on lysine in histone, the electrostatic interaction between histone and the negatively charged DNA is weakened, followed by neutralization of the lysine positive charge. Consequently, the euchromatin loosens to maintain a more open status, thereby increasing the ability of DNA replication or transcription due to nucleosome conformation change [Citation1]. Conversely, inhibition of acetylation by histone deacetylase (HDAC) can cause transcriptional silencing [Citation2]. Appropriate histone acetylation is known to be important for various physiological cellular processes, whereas aberrant HAT or HDAC-induced abnormal histone acetylation is closely related to the development of cancers. Previous studies have shown that high expression of HDAC is associated with poor prognosis in gastric, ovarian, and oesophageal cancers [Citation3,Citation4]. Abnormal histone acetylation is involved in the silencing of tumour suppressor gene CDH1, cell cycle-related genes p21and p27, pro-apoptotic genes Bax and AIF, etc. [Citation5]. Therefore, HDAC inhibition or HAT activation might facilitate anti-tumour potentiation by restoring the normal expression of tumour suppressor genes.
In recent years, the histone acetylation targeted therapy has been focused on HDAC inhibitors (HDACis). To date, several HDACis, such as vorinostat, belinostat and romidepsin, have been successful in the treatment of haematological malignancies [Citation6]. As the first HDACi approved by the Food and Drug Administration (FDA), vorinostat was reported to successfully treat refractory cutaneous T-cell lymphoma (CTCL) in phase II clinical trials, which extended the longest survival time to 23 weeks [Citation7]. Another HDAC inhibitor 4-Phenylbutyrate acid (4-PBA), which is also known as the orphan drug for urea cycle disorders, was also reported to be able to inhibit cell proliferation by inducing apoptosis, cell cycle arrest, and senescence in colon, gastric and breast cancers [Citation8–Citation10]. Moreover, the safety of the salt of 4-PBA was also confirmed in the phase I and phase II clinical trials of haematological malignancies, colon carcinoma, anaplastic astrocytoma, etc. [Citation8–Citation11]. However, most clinical trials with HDACis, including 4-PBA, in solid tumours showed no obvious benefits [Citation9,Citation11]. The possible reasons were thought that the pharmacokinetic properties of HDACi might limit their distribution in solid tumours, or the role of HDACi in solid tumours might be more complicated than in haematological malignancies. Therefore, to develop effective HDACis for solid tumours, it is critical to clarify their role and underlying mechanisms in solid tumours.
Our previous study showed that 4-PBA could promote cell migration by upregulating HER3/HER4 expression in gastric cancer. In contrast, another study reported that 4-PBA inhibited the migration of metastatic prostate cancer cells [Citation12]. In addition, 4-PBA had no effect on the cell migration in colorectal cancer [Citation13]. Similar contradictory results were also reported for another HDACi valproic acid (VPA), which was shown to have opposite effects on epithelial–mesenchymal transition (EMT) in hepatocarcinoma and cholangiocarcinoma cells [Citation14,Citation15]. Taken together, these studies suggested that HDACis play different roles in different tumours, and HDACi-promoted migration might be a reason for their failure in solid tumour treatment.
Given the high incidence of cancer in Asia, especially in China, most patients of gastric cancer are diagnosed at an advanced stage. Chemotherapy, targeting and other drugs are the main treatments for gastric cancer, but the efficacy is limited. Hence, it is necessary to develop new therapeutic strategies. Although HDACis were unsuccessful in the treatment of solid tumours, they showed anti-cancer potential in various cancers including gastric cancer, suggesting that HDACis have the potential to become an epigenetic drug candidate for cancer. Therefore, comprehensively investigating the effect and related mechanism of HDACis on gastric cancer cells would help to develop more effective drugs for gastric cancer.
Based on our previous findings that 4-PBA could promote gastric cancer cell migration, we investigated the detailed mechanisms of 4-PBA on gastric cancer cells in this study. The results showed that high-concentration of 4-PBA inhibited cell proliferation, while low concentration promoted metastasis of gastric cancer cells mediated by IL-8 upregulation followed by activation of the Gab2-ERK pathway. We speculated that this might be a possible mechanism for failure of 4-PBA in solid tumour therapy. This study provided a new comprehensive understanding about HDACi mechanisms in the therapy of solid tumours.
Results
4-PBA induced atypical EMT with aberrant E-cadherin expression in gastric cancer cells
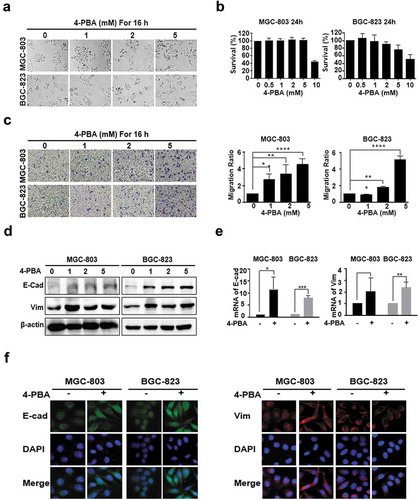
After treatment with 0, 0.5, 1, 2, 5 and 10 mM of 4-PBA for 16 h, the morphology of the MGC-803 and BGC-823 cells changed from an epithelial sheet-like structure to a mesenchymal fibroblastic spindle shape in a dose-dependent manner ()). No significant cytotoxicity was detected at concentrations under 5 mM ()), indicating that low doses of 4-PBA might induce EMT in gastric cancer. The results of the transwell assay showed that 5 mM 4-PBA treatment enhanced the rate of migration to 451.7 ± 39.8% and 512 ± 26% in MGC-803 and BGC-823 cells, respectively ()). However, both the epithelial marker E-cadherin and the mesenchymal marker vimentin were significantly upregulated by 4-PBA at protein and mRNA levels (, )), which was in conflict with classical EMT characterized by E-cadherin downregulation and vimentin upregulation [Citation16,Citation17]. Hence, the localization of E-cadherin and vimentin was further observed by immunofluorescence. As shown in ), vimentin accumulated in the cytoplasm after 4-PBA-treatment, which was consistent with previous studies, whereas E-cadherin was significantly increased in the cytoplasm rather than on the membrane, indicating its ectopic expression induced by 4-PBA. These results strongly suggested that 4-PBA induced an atypical EMT with aberrant E-cadherin expression.
Figure 1. 4-PBA induced gastric cancer cells EMT with aberrant E-cadherin expression.
4-PBA induced EMT by upregulating IL-8 in gastric cancer cells
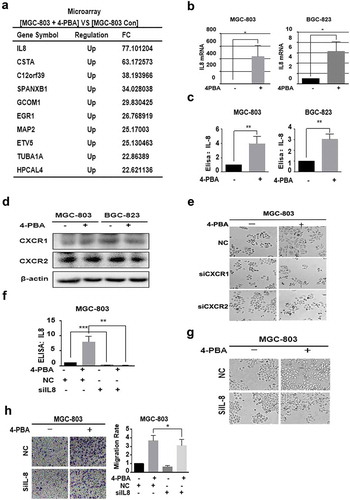
To investigate the mechanisms of 4-PBA in EMT induction in gastric cancer cells, Affymetrix GeneChip Human Transcriptome Array 2.0 analysis was performed. Among the 4-PBA-upregulated genes ()), IL-8 was the most significantly upregulated by 4-PBA. Hence, it was selected for further investigation of its involvement in 4-PBA-induced EMT. The changes in IL-8 mRNA levels by 4-PBA treatment were consistent with the result of microarray analysis in MGC-803 and BGC-823 cells ()). Similar, the secretion level of IL-8 was also increased 4-PBA in gastric cancer cells ()). Since we speculated that 4-PBA-induced IL-8 expression might promote migration by autocrine process, the effect of IL-8 receptors, CXCR1 and CXCR2, on 4-PBA-induced EMT was then investigated. It was shown that CXCR1 and CXCR2 were expressed in both of two gastric cancer cell lines ()), and silencing CXCR1 or CXCR2 partial reversed the EMT morphology induced by 4-PBA in MGC-803 cells, further confirming our conjecture ()). Then, IL-8 involvement in 4-PBA-induced EMT was investigated. When IL-8 expression was silenced by siRNA ()), the migration ability and EMT morphologies induced by 4-PBA were partially reversed in both cells (, )). These data strongly suggested that 4-PBA induced gastric cancer cell EMT by up-regulating IL-8.
Figure 2. 4-PBA-induced EMT by IL-8 upregulation in gastric cancer cells.
IL-8 upregulated by 4-PBA induced EMT through the ERK signalling pathway in gastric cancer cells
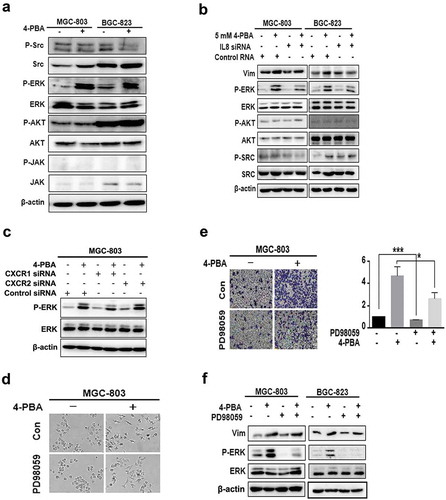
Next, to investigate the mechanisms of IL-8-mediated 4-PBA-induced EMT, the activation of AKT, ERK, JAK and SRC, the downstream molecules of IL-8, was detected by Western blot. ) shows that the phosphorylation level of ERK was increased by 4-PBA treatment, which was partially reversed by knockdown of IL-8 or CXCR1 or CXCR2 (, )). Similarly, treatment with ERK inhibitor PD98059 led to partial reversal of the EMT morphologies, cell migration and vimentin upregulation induced by 4-PBA (, , )). These data suggested that 4-PBA induced-EMT was mediated by IL-8-activated ERK pathway in gastric cancer cells.
Figure 3. 4-PBA-upregulated IL-8 induced EMT through ERK signalling pathway in gastric cancer cells.
IL-8 activated ERK signalling pathway via Gab2 in gastric cancer cells
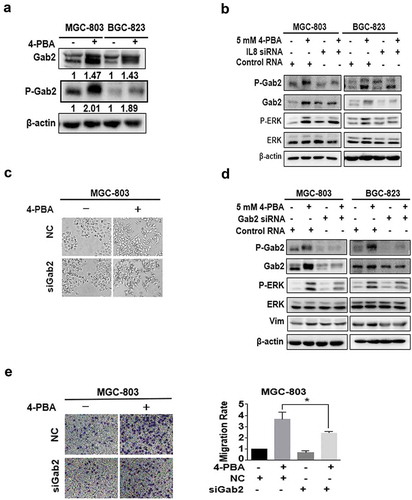
IL-8 was known to activate downstream signalling through the binding of CXCR1/CXCR2 with Gab2, the docking protein, and subsequent phosphorylation of Gab2 [Citation18,Citation19]. Therefore, whether Gab2 participates in 4-PBA-induced-IL-8 pathway was examined. The results showed that 4-PBA enhanced the phosphorylation levels of Gab2 ()), which was partially inhibited by IL-8 knockdown in gastric cancer cells, along with the downstream ERK ()). Moreover, Gab2 knockdown partially reversed 4-PBA-induced EMT morphology ()) and migration ()), inhibited the activation of ERK and the upregulation of vimentin ()). Taken together, these results suggested that 4-PBA induced EMT through IL-8-Gab2-ERK signalling pathway in gastric cancer cells.
Figure 4. Gab2 phosphorylation mediated IL-8-induced ERK activation.
4-PBA promoted IL-8 gene transcription via enhancing the histone acetylation level in IL-8 promoter regions
As an HDAC inhibitor, 4-PBA can promote gene transcription by enhancing histone acetylation. Therefore, we speculated that 4-PBA might upregulate IL-8 by inhibiting HDAC activity and enhancing histone acetylation. First, the levels of histones H2A, H2B, H3 and H4 were investigated after exposure to 4-PBA in MGC-803 and BGC-823 cells. The levels of Ace-H2AK5, Ace-H2BK5, Ace-H3K9 and Ace-H4K8, especially Ace-H3K9, were increased ()), indicating that the acetylation of H3 might play an important role in promoting IL-8 gene transcription. The changes in acetylation levels in other H3 loci, including K14, K18, K27 and K56 were further detected. As expected, all H3 loci were obviously enhanced ()). The HDAC activity was also inhibited after treatment with 4-PBA ()). These results indicated that 4-PBA increased the acetylation level of histones, especially H3, by inhibiting the HDAC activity. Furthermore, whether 4-PBA could promote the transcription activity of IL-8 was investigated using CHIP assay. The results showed that 4-PBA significantly increased the binding of acetyl-Histone3 (H3ac) and IL-8 ()). These results strongly suggested that 4-PBA promoted IL-8 transcription by inhibiting HDAC activity and enhancing histone acetylation.
Figure 5. 4-PBA upregulated IL-8 expression and IL-8 pathway activation through HDAC activity inhibition.
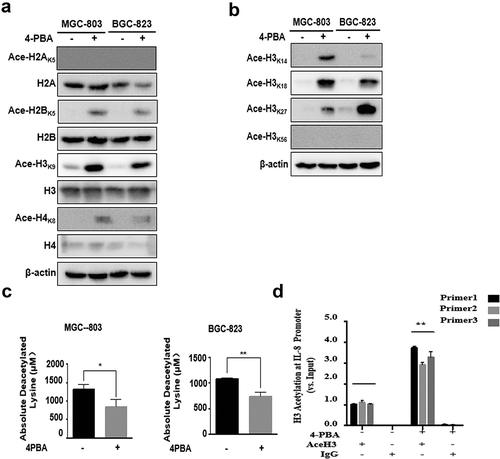
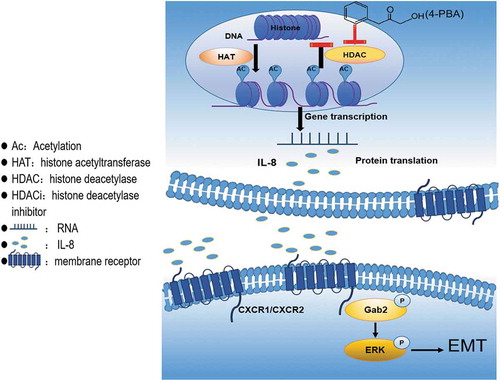
Figure 6. Schematic representation of the proposed model.
Discussion
In this study, we found that although 4-PBA could inhibit cell proliferation at high concentrations, low concentrations of 4-PBA (under 5mM) could induce EMT and promote cell migration by H3 acetylation-mediated upregulation of IL-8 and the activation of downstream Gab2-ERK pathway in gastric cancer cells ( was the schematic representation of the proposed model), which might partially explain why HDACis failed in solid tumour therapy.
EMT is known to play an important role in tumour cell progression and metastasis. We found that 4-PBA could induce EMT-like morphology and promote cell migration in a dose-dependent manner under 5mM in gastric cancer, which is similar to previous studies that SAHA were reported to be able to induce EMT in ovarian cancer cells [Citation20]. However, as compared to classical EMT with the characteristics of epithelial marker E-cadherin downregulation and mesenchymal marker vimentin upregulation [Citation21], we found that both E-cadherin and vimentin were significantly upregulated by 4-PBA in this study. This change was different from the previous report that although TSA (Trichostatin A) induced mesenchymal-like morphological changes with E-cadherin upregulation, the migration was inhibited in human gastric cancer cells and breast cancer cells [Citation22]. Immunofluorescence study showed that E-cadherin was mainly localized in the cytoplasm but not on the membrane, suggesting its abnormal expression. Previous studies reported that high expression of cytoplasmic E-cadherin in gastric cancer cells was associated with the depth of infiltration and regional lymph node metastasis [Citation23], as well as was an independent predictor in bladder urothelial carcinoma to reduce the disease-free interval/time to recurrence [Citation24]. Therefore, 4- PBA- induced EMT with abnormal E-cadherin expression might lead to poor therapeutic effect in gastric cancer. It is unclear how 4-PBA upregulated E-cadherin with abnormal localization. HDACi MS-275 was reported to increase the correct variant transcript splicing and decrease the exon 11-skipped aberrant transcript through promoting histone acetylation of the promoter region in chronic lymphocytic leukaemia (CLL) [Citation25]. Whether 4-PBA could affect E-cadherin re-expression and location by gene transcription splicing needs further investigation.
IL-8, the most significantly up-regulated gene by 4-PBA, was found to induce EMT in gastric cancer cells. IL-8, also known as CXCL8, is involved in inflammation response and tumour development [Citation26–Citation29]. TSA, another HDACi was also reported to increase the expression of IL-8 in HeLa cells and breast cancer cells, which was similar to our results. IL-8 upregulation was mediated by HDACi-induced expression of IκB kinase (IKK) in breast cancer [Citation30–Citation32]. However, in this study, 4-PBA directly promoted the acetylation of the promoter region and thus up-regulated the expression of IL-8. In addition to the role of IL-8 in cell proliferation and angiogenesis in prostate cancer [Citation33], recent evidence showed that IL-8 participated in the EMT process in colorectal cancer cells [Citation34–Citation36]. Stable overexpression of exogenous IL-8 was shown to facilitate EMT in thyroid cancer [Citation36]. In addition, SNAIL induced EMT accompanied by the upregulation of IL-8, which was reversed by IL-8 knockdown in human colorectal carcinoma [Citation35]. In this study, we revealed that 4-PBA-mediated IL-8 upregulation played an important role in 4-PBA-induced EMT in gastric cancer cells.
Gab2 is the classical downstream signalling factor of IL-8-CXCR1/CXCR2 [Citation19]. Gab2 is an important signal transducer of various extracellular stimuli, such as growth factors, cytokines and antigen receptors, and connects the receptors and intracellular downstream effectors to facilitate protein–protein interactions. Gab2 can promote cell growth, differentiation, migration and apoptosis by triggering the activation of downstream factors [Citation37]. In this study, after treatment with 4-PBA, both the protein and phosphorylation levels of Gab2 were increased, whereas knockdown of IL-8 partially inhibited the activation of Gab2 and downstream ERK, suggesting that upregulation of IL-8 by 4-PBA induced gastric cancer cell EMT mediated by the Gab2-ERK pathway. However, we also noticed that the protein level of Gab2 was upregulated with 4-PBA-treatment, which was only partially suppressed by IL-8 knockdown, indicating that 4-PBA might also upregulate Gab2 expression by inhibiting HDAC activity. Therefore, in this study, Gab2 might be activated not only by 4-PBA-upregulated IL-8, but also by 4-PBA directly upregulated Gab2 itself. Further study needs to investigate the mechanism of Gab2 up-regulation and the role of Gab2 in 4-PBA-induced EMT. Knockdown of IL-8 partially inhibited the activation of Gab2 and downstream ERK, suggesting that upregulation of IL-8 by 4-PBA induced gastric cancer cell EMT mediated by the Gab2-ERK pathway. Therefore, our results also indicated that combining Gab2 inhibitor with 4-PBA might enhance the anti-tumour effect of 4-PBA by attenuating its EMT induction role.
In summary, this is the first study to demonstrate that 4-PBA induced gastric cancer cell EMT by inhibiting HDAC and enhancing histone acetylation. The activation of IL-8-Gab2-ERK pathway played an important role in this process. These findings may facilitate a new understanding of innovative tumour treatments using 4-PBA. The possible potential risks of cancer progression should be carefully monitored when using 4-PBA to treat cancer patients.
Materials and methods
Cells and cell culture
Human gastric cell lines MGC-803 and BGC-823 obtained from the Type Culture Collection of the Chinese Academy of Sciences (China). Cells were maintained in RPMI 1640 medium (Rosewell Park Memorial Institute) with 10% foetal bovine serum (FBS) and 1% penicillin–streptomycin in a humidified atmosphere of 95% air and 5% CO2 at 37°C. Cells were sub-cultured every 2–3 days and harvested in their logarithmic phase of growth.
Reagents and antibodies
4-PBA was purchased from Sigma (USA). The ERK inhibitor PD98059 was obtained from Promega (Madison, WI, USA). Rabbit anti-β-actin (sc-16-16-R), anti-ERK1/2 (sc-292,838) antibodies were obtained from Santa Cruz Biotechnology; Mouse, anti-Src (8056), anti-IL-8 (376,750), were obtained from Santa Cruz Biotechnology; Rabbit, anti-E-cadherin (3195s), anti-vimentin (5741s), anti-p-Stat3 (9145s), anti-Stat3 (4904s), anti-p-Src (6943s), anti-AKT (9272s), anti-p-AKT (Ser473), anti-p-Gab2 (3882), anti-p-ERK1/2 (Thr202/Tyr204), anti-Acetyl-Histone3, anti-Histone2A (2576), anti-Histone2B (12,349), anti- Histone3 (4499), anti-Histone4 (13,919), anti-Acetyl-H2AK5 (2576), anti-Acetyl-H2BK5 (12,799), anti-Acetyl-H3K9 (9649), anti-Acetyl-H4K8 (2594), anti-Acetyl-H3K14 (7627), anti-Acetyl-H3K18 (13,998), anti-Acetyl-H3K27 (8173), anti-Acetyl-H3K56 (4243), anti-α/β-Tublin (2148s), anti-Acetyl-α-Tublin (5335s), anti-HDAC4 (7628), anti-phospho-HDAC4(Ser246)/HDAC5(Ser259)/HDAC7(Ser155) (3443), anti-HDAC6 (7558) antibodies were purchased from Cell Signalling Technology; Mouse anti-HDAC1 (5356), anti-HDAC2 (5113), anti-HDAC3 (3949) antibodies were purchased from Cell Signalling Technology;Mouse anti-CXCL8/IL-8 neutralization (MAB208-100) was purchased from R&D; Mouse anti-CXCR1 (ab195985) and rabbit anti-CXCR2 (ab65968) antibodies were purchased from Abcam; Rabbit anti-Gab2 (06–967) was purchased from Millipore.
Small interfering RNA transfections
The MGC-803 cells were seeded at 1.2 × 105 per well in 6-well plates. After incubated overnight, the cells were transfected with siRNAs using Lipofectamine 3000 (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions. siRNA sequences (Genepharma) for IL-8 and Gab2 were as follows: Gab2 (5ʹGAGACAGCG AAGAGAACUA-3ʹ). The negative control (5ʹ-AATTCTCCGAACGTGTCACGT-3ʹ). After 48 h of transient transfection, the cells were treated with 4-PBA for 16 h. The level of protein change was verified by western blot. The effect of IL-8, and Gab2 genes on migration was detected by transwell assay.
Western blot
Cells were seeded at 2 × 105 per well in 6-well plates and incubated overnight; Cells were treated with 4-PBA (5 mM) for indicated times. Cells were lysed in lysis buffer. The method of Western blot was discussed in our previous study [Citation38].
Migration assay
Migration assays were carried out in the 24-well chamber and polycarbonate inserts with 8-μm pore size membranes (Corning, USA). The cells (3 × 104 cells/well) pretreated with 4-PBA (1, 2, 5 mM), PD98059 (25 μM) or trasnsfed with siRNA were loaded into the upper chamber with 200μl serum-free RPMI 1640 medium. The lower chambers contained 500μl of RPMI 1640 with 2.5% FBS. After incubation for 24 h, the migrated cells onto the outer side of the membrane were stained with 0.1% Giemsa solution for 1 h. Five different fields were captured and counted at ×20 magnification per well.
Chromatin immunoprecipitation (ChIP)
MGC-803 cells were treated with or without 4-PBA in 10 cm dishes. ChIP protocol was from CHIP-IT Express Enzymatic Magnetic Chromatin Immunoprecipitation Kit & Enzymatic Shearing Kit (Catalogue No. 53,009, Active Motif North America). DNA was sheared into ~200 bp pieces with enzyme. After removing aliquots of each for input controls, the remainder was immunoprecipitated with anti-acetyl-Histone3 (06–599 Millipore), anti-acetyl-Histone4 (06–866 Millipore), anti-IgG (3900s) as control antibodies. Quantitative Real-Time PCR of precipitated DNA described below.
Quantitative real-time PCR
MGC-803 and BGC-823 cells were treated with 4-PBA for 24 h, and then washed twice with ice-cold PBS. Total mRNA was extracted with TRIZOL reagent. The cDNA was generated from 800 ng total RNA using SYBR® Premix EX TaqTM II (Tli RNaseH Plus, Takara, Japan). Precipitated DNAs were purificated with ChIP DNA Purification Kit (Catalogue Nos. 58,002, Active Motif North America). Quantitative Real-TimePCR was run on Applied Biosystems® 7500 Real-Time PCR Systems (Thermo Fisher SCIENTIFIC, USA) using validated primers of 18S, IL-8, E- cadherin, vimentin and SYBR Premix Ex Taq II (Takara, Japan) for detection. The cycle number when the fluorescence first reaches a preset threshold (Ct) allow the quantification of the specific template concentration. Transcripts of 18S in the same incubations were used as internal control. Primer sequences for IL-8: Forward (5ʹ-AAGCTGGCCGTGGCTCTCT T-3ʹ), Reverse (5ʹ-TGGTGGCGCAGTGTGGTCCA-3ʹ). Primer sequences for E-cadherin: Forward (5ʹ-GAAGAGAGACTGGGTTATTCCTCCC-3ʹ), Reverse (5ʹ-CAGTGATGCTGTAGAAAACCTTGCC-3ʹ). Primer sequences for vimentin: Forward (5ʹ-GCACGATGAGGAATCCAGGA-3ʹ), Reverse (5ʹ-AGGTCAGGCTTGG AAACATCC-3ʹ). Primer sequences for 18S: Forward (5ʹ-CCCGGGGAGGTAGTGACG AAAAAT-3ʹ), Reverse (5ʹ-CGCCCGCCCGCTCCCAAGAT- 3ʹ).
Immunofluorescence
The cells were seeded in Lab-Tek chamber slides (Nunc S/A, Polylabo, Strasbourg, France). After seeded overnight, the cells were treated with or without 4-PBA for 16 h and fixed in 3.3% paraformaldehyde for 15 min, then permeabilized with 0.2% Triton X-100 for 5 min, blocked with 5% bovine serum albumin (BSA) for 1 h and then incubated with anti-E- cadherin and anti-vimentin antibody overnight at 4°C. The next day, Alexa Fluor 546-conjugated goat anti-rabbit IgG or Alexa Fluor 488-conjugated goat anti-rabbit IgG (Molecular Probes) were added in blocking solution for 1 h at room temperature in the dark. 4′6′-diamidino-2-phenylindole (DAPI) was used to stain nuclei for 5 min. After mounted with the Slow Fade Antifade Kit (Molecular Probes, Eugene, OR, USA), the cells were visualized by fluorescence microscopy (BX53, Olympus, Japan).
Statistical analysis
All the experimental data were expressed as means ± standard deviation and the mean values were calculated from at least three independent experiments. Statistical comparisons were calculated by Student’s two-tailed t-test. P < 0.05 was considered as statistically significant.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Dawson MA, Kouzarides T. Cancer epigenetics: from mechanism to therapy. Cell. 2012 Jul 6;150(1):12–27. PubMed PMID: 22770212; eng.
- Menzies KJ, Zhang H, Katsyuba E, et al. Protein acetylation in metabolism – metabolites and cofactors. Nat Rev Endocrinol. 2016 Jan;12(1):43–60. PubMed PMID: 26503676; eng.
- Weichert W, Denkert C, Noske A, et al. Expression of class I histone deacetylases indicates poor prognosis in endometrioid subtypes of ovarian and endometrial carcinomas. Neoplasia (New York, NY). 2008 Sep;10(9):1021–1027. PubMed PMID: 18714364; PubMed Central PMCID: PMCPmc2517648. eng.
- Weichert W, Roske A, Gekeler V, et al. Association of patterns of class I histone deacetylase expression with patient prognosis in gastric cancer: a retrospective analysis. Lancet Oncol. 2008 Feb;9(2):139–148. PubMed PMID: 18207460; eng.
- Jia D, Augert A, Kim DW, et al. Crebbp loss drives small cell lung cancer and increases sensitivity to HDAC inhibition. Cancer Discov. 2018 Nov;8(11):1422–1437. PubMed PMID: 30181244; PubMed Central PMCID: PMCPmc6294438. eng.
- Mottamal M, Zheng S, Huang TL, et al. Histone deacetylase inhibitors in clinical studies as templates for new anticancer agents. Molecules. 2015 Mar 2;20(3):3898–3941. PubMed PMID: 25738536; PubMed Central PMCID: PMCPmc4372801. eng.
- Iwamoto M, Friedman EJ, Sandhu P, et al. Clinical pharmacology profile of vorinostat, a histone deacetylase inhibitor. Cancer Chemother Pharmacol. 2013 Sep;72(3):493–508. PubMed PMID: 23820962; eng.
- Carducci MA, Gilbert J, Bowling MK, et al. A phase I clinical and pharmacological evaluation of sodium phenylbutyrate on an 120-h infusion schedule. Clin Cancer Res off J Am Assoc cancer res. 2001 Oct;7(10):3047–3055. PubMed PMID: 11595694; eng.
- Lin J, Gilbert J, Rudek MA, et al. A phase I dose-finding study of 5-azacytidine in combination with sodium phenylbutyrate in patients with refractory solid tumors. Clin Cancer Res off J Am Assoc cancer res. 2009 Oct 1;15(19):6241–6249. 10.1158/1078-0432.ccr-09-0567. PubMed PMID: 19789320; PubMed Central PMCID: PMCPmc2845396. eng.
- Maslak P, Chanel S, Camacho LH, et al. Pilot study of combination transcriptional modulation therapy with sodium phenylbutyrate and 5-azacytidine in patients with acute myeloid leukemia or myelodysplastic syndrome. Leukemia. 2006 Feb;20(2):212–217. PubMed PMID: 16357841; eng.
- Camacho LH, Olson J, Tong WP, et al. Phase I dose escalation clinical trial of phenylbutyrate sodium administered twice daily to patients with advanced solid tumors. Invest New Drugs. 2007 Apr;25(2):131–138. PubMed PMID: 17053987; eng.
- McAuley EM, Bradke TA, Plopper GE. Phenylboronic acid is a more potent inhibitor than boric acid of key signaling networks involved in cancer cell migration. Cell Adh Migr. 2011 September–-October;5(5):382–386. PubMed PMID: 21975546; PubMed Central PMCID: PMCPmc3218604. eng.
- Lv LV, Zhou J, Lin C, et al. DNA methylation is involved in the aberrant expression of miR-133b in colorectal cancer cells. Oncol Lett. 2015 Aug;10(2):907–912. PubMed PMID: 26622593; PubMed Central PMCID: PMCPmc4509424. eng.
- Wu L, Feng H, Hu J, et al. Valproic acid (VPA) promotes the epithelial mesenchymal transition of hepatocarcinoma cells via transcriptional and post-transcriptional up regulation of snail. Biomed Pharmacothe. 2016 Dec;84:1029–1035. PubMed PMID: 27768928; eng.
- Wang JH, Lee EJ, Ji M, et al. HDAC inhibitors, trichostatin A and valproic acid, increase ecadherin and vimentin expression but inhibit migration and invasion of cholangiocarcinoma cells. Oncol Rep. 2018 Jul;40(1):346–354. PubMed PMID: 29767267; eng.
- Yilmaz M, Christofori G. EMT, the cytoskeleton, and cancer cell invasion. Cancer Metastasis Rev. 2009 Jun;28(1–2):15–33. PubMed PMID: 19169796; eng.
- Huang RY, Guilford P, Thiery JP. Early events in cell adhesion and polarity during epithelial-mesenchymal transition. J Cell Sci. 2012 Oct 1;125(Pt 19):4417–4422. PubMed PMID: 23165231; eng.
- Neel BG, Gu H, Pao L. The ‘Shp’ing news: SH2 domain-containing tyrosine phosphatases in cell signaling. Trends Biochem Sci. 2003 Jun;28(6):284–293. PubMed PMID: 12826400; eng.
- Gu H, Pratt JC, Burakoff SJ, et al. Cloning of p97/Gab2, the major SHP2-binding protein in hematopoietic cells, reveals a novel pathway for cytokine-induced gene activation. Mol Cell. 1998 Dec;2(6):729–740. PubMed PMID: 9885561; eng.
- Tang HM, Kuay KT, Koh PF, et al. An epithelial marker promoter induction screen identifies histone deacetylase inhibitors to restore epithelial differentiation and abolishes anchorage independence growth in cancers. Cell Death Discov. 2016;2:16041. PubMed PMID: 27551531; PubMed Central PMCID: PMCPmc4979427. eng.
- Ying R, Wang XQ, Yang Y, et al. Hydrogen sulfide suppresses endoplasmic reticulum stress-induced endothelial-to-mesenchymal transition through Src pathway. Life Sci. 2016 Jan;1(144):208–217. PubMed PMID: 26656263; eng.
- Han RF, Li K, Yang ZS, et al. Trichostatin A induces mesenchymal-like morphological change and gene expression but inhibits migration and colony formation in human cancer cells. Mol Med Rep. 2014 Dec;10(6):3211–3216. PubMed PMID: 25269990; eng.
- Guzman P, Araya J, Villaseca M, et al. Immunohistochemical expression of the E-cadherin-catenin complex in gastric cancer. Revista Medica de Chile. 2006 Aug;134(8):1002–1009. PubMed PMID: 17130988; spa.
- Gallagher EM, O’Shea DM, Fitzpatrick P, et al. Recurrence of urothelial carcinoma of the bladder: a role for insulin-like growth factor-II loss of imprinting and cytoplasmic E-cadherin immunolocalization. Clin Cancer Res off J Am Assoc cancer res. 2008 Nov 1;14(21):6829–6838. PubMed PMID: 18980977; eng.
- Jordaan G, Liao W, Sharma S. E-cadherin gene re-expression in chronic lymphocytic leukemia cells by HDAC inhibitors. BMC Cancer. 2013 Feb 25;13:88. PubMed PMID: 23432814; PubMed Central PMCID: PMCPmc3586366. eng.
- Waugh DJ, Wilson C. The interleukin-8 pathway in cancer. Clin Cancer Res off J Am Assoc cancer res. 2008 Nov 1;14(21):6735–6741. PubMed PMID: 18980965; eng.
- Balkwill F, Mantovani A. Inflammation and cancer: back to Virchow? Lancet. 2001 Feb 17;357(9255):539–545. PubMed PMID: 11229684; eng.
- Holmes WE, Lee J, Kuang WJ, et al. Structure and functional expression of a human interleukin-8 receptor. Science (New York, NY). 1991 Sep 13;253(5025):1278–1280. PubMed PMID: 1840701; eng.
- Araki S, Omori Y, Lyn D, et al. Interleukin-8 is a molecular determinant of androgen independence and progression in prostate cancer. Cancer Res. 2007 Jul 15;67(14):6854–6862. PubMed PMID: 17638896; eng.
- Vancurova I, Uddin MM, Zou Y, et al. Combination therapies targeting HDAC and IKK in solid tumors. Trends Pharmacol Sci. 2018 Mar;39(3):295–306. PubMed PMID: 29233541; PubMed Central PMCID: PMCPmc5818305. eng.
- Adam E, Quivy V, Bex F, et al. Potentiation of tumor necrosis factor-induced NF-kappa B activation by deacetylase inhibitors is associated with a delayed cytoplasmic reappearance of I kappa B alpha. Mol Cell Biol. 2003 Sep;23(17):6200–6209. PubMed PMID: 12917341; PubMed Central PMCID: PMCPmc180966. eng.
- Chavey C, Muhlbauer M, Bossard C, et al. Interleukin-8 expression is regulated by histone deacetylases through the nuclear factor-kappaB pathway in breast cancer. Mol Pharmacol. 2008 Nov;74(5):1359–1366. PubMed PMID: 18669446; eng.
- Brat DJ, Bellail AC, Van Meir EG. The role of interleukin-8 and its receptors in gliomagenesis and tumoral angiogenesis. Neuro Oncol. 2005 Apr;7(2):122–133. PubMed PMID: 15831231; PubMed Central PMCID: PMCPmc1871893. eng.
- Cheng XS, Li YF, Tan J, et al. CCL20 and CXCL8 synergize to promote progression and poor survival outcome in patients with colorectal cancer by collaborative induction of the epithelial-mesenchymal transition. Cancer Lett. 2014 Jun 28;348(1–2):77–87. PubMed PMID: 24657657; eng.
- Hwang WL, Yang MH, Tsai ML, et al. SNAIL regulates interleukin-8 expression, stem cell-like activity, and tumorigenicity of human colorectal carcinoma cells. Gastroenterology. 2011 Jul;141(1):279–91, 291.e1–5. PubMed PMID: 21640118; eng.
- Visciano C, Liotti F, Prevete N, et al. Mast cells induce epithelial-to-mesenchymal transition and stem cell features in human thyroid cancer cells through an IL-8-Akt-Slug pathway. Oncogene. 2015 Oct 1;34(40):5175–5186. PubMed PMID: 25619830; eng.
- Ding CB, Yu WN, Feng JH, et al. Structure and function of Gab2 and its role in cancer (Review). Mol Med Rep. 2015 Sep;12(3):4007–4014. PubMed PMID: 26095858; PubMed Central PMCID: PMCPmc4526075. eng.
- Li H, Xu L, Zhao L, et al. Insulin-like growth factor-I induces epithelial to mesenchymal transition via GSK-3beta and ZEB2 in the BGC-823 gastric cancer cell line. Oncol Lett. 2015 Jan;9(1):143–148. PubMed PMID: 25435948; PubMed Central PMCID: PMCPmc4246767. eng.