
ABSTRACT
In mammals, DNA methylation patterns are established by various types of DNA methyltransferases and can be stably passed on during cell division, thus creating a paradigm for epigenetic regulation that can mediate long-lasting changes in gene expression even when the initial triggering signal has disappeared. Although functional deficiency of DNMT3A, one of the methyltransferases, leads to abnormal DNA methylation patterns that result in developmental deficits in mammals, the impacts of its overexpression on tissue gene expression and DNA methylation patterns remain unclear. Here, our previously established hDNMT3A transgenic rat model and mRNA sequencing and bisulphite sequencing PCR were used to analyse the impact of hDNMT3A overexpression on tissue transcriptome and methylome, and whether the impact could be inherited intergenerationally was subsequently investigated. Our results revealed that the overexpression of hDNMT3A could induce notable gene expression variations in rat testis and brain. More importantly, 36.02% and 38.89% of these variations could be intergenerationally inherited to offspring without the transmission of the initial endogenic trigger in the brain and testis, respectively. Furthermore, we found that intergenerationally inherited DNA methylation variations in their promoters and exons could be the underlying mechanism. Compared with inheritable variations that were passively induced by environmental factors, these variations were actively induced by endogenous epigenetic modifiers. This study provided evidence for the epigenetic inheritance of endogenous factors that actively induce gene expression and DNA methylation variations; however, more studies are needed to determine the number of generations that these variations can be stably inherited.
Introduction
DNA methylation at the 5-position of cytosine (5mC) on CpG dinucleotides is the most common epigenetic modification in mammals, and it represents a heritable marker and plays important roles in regulating gene expression, controlling monoallelic expression of genes during imprinting, silencing transposons and repeat elements, inactivating X-chromosomes and reorganizing chromatin [Citation1]. In the mammalian genome, the majority of CpGs (70–80%) are methylated except in gene regulatory regions, notably promoters, CpG islands and large undermethylated regions termed canyons, where unmethylated CpGs are often clustered [Citation2]. These patterns of DNA methylation are established by various types of DNA methyltransferases (DNMTs) and can be stably passed on during cell division, thus creating a paradigm for epigenetic regulation that can mediate long-lasting changes in gene expression even when the initial triggering signal has disappeared [Citation3]. To date, six different DNMTs have been functionally and structurally characterized in mammals, including four active DNMTs, DNMT1, DNMT3A, DNMT3B and DNMT3C [Citation4,Citation5]; one catalytically inactive member, DNMT3L; and one RNA methyltransferase, DNMT2 [Citation6]. While DNMT3A, DNMT3B and DNMT3C establish patterns of DNA methylation through de novo methylation pathways, DNMT1 primarily functions in maintenance methylation during DNA replication, and DNMT3 L acts as a cofactor for DNMT3A [Citation4]. DNMT3A and DNMT3B have been reported to be essential for genome-wide de novo methylation during embryo implantation and primordial germ cell (PGC) development to establish new methylation patterns in offspring embryos and germlines, respectively [Citation7].
Previous studies have reported that abnormal activity of these DNMTs can induce aberrant DNA methylation patterns, resulting in gene expression disorders and leading to the occurrence of disease. For example, knockout of DNMT3A or DNMT3B causes embryonic lethality in mice [Citation7]; mutation of R882H in DNMT3A results in methylation loss and leads to acute myeloid leukaemia and Tatten-Brown-Rahman syndrome in humans [Citation8]; overexpression of DNMT1, DNMT3A and DNMT3B is correlated with tumorigenesis and other diseases [Citation9–Citation11]; and expression patterns of DNMT3A are significantly associated with human gastric and hepatocellular carcinoma prognoses [Citation12,Citation13]. In the adult brain, DNMT1 and DNMT3A are highly expressed in postmitotic neurons [Citation14]. In mammalian testis, DNMTs play important roles in gametogenesis, and conditional DNMT3A knockout in germ cells impairs spermatogenesis and is associated with methylation loss of paternally imprinted genes [Citation15]. Although functional deficiency of DNMTs leads to abnormal DNA methylation patterns that result in disease in humans and developmental deficits in mammals in general, the impacts of DNMT overexpression on tissue gene expression patterns remain unclear.
In mice, conditional overexpression of the human-derived histone 3 lysine 4 (H3K4) demethylase hKDM1A (another epigenetic modifier) reduces H3K4 dimethylation and alters RNA content in mouse sperm [Citation16], and its overexpression affects the embryo development and survive rate of the offspring, and the impact could be transmitted to the F3 generation without the transmission of the hKDM1A transgene. In our previous study, we established human-derived DNMT3A (hDNMT3A)-transgenic rats and found that hDNMT3A was highly expressed in the brain and testis [Citation17]. Furthermore, its overexpression in rat testis induced intergenerational inheritance of DNA methylation variations in mature sperm [Citation17]. However, whether hDNMT3A overexpression influenced the transcriptome of the rat brain and testis and whether these affected gene expression patterns could be inherited to the offspring tissues without the transmission of the hDNMT3A transgene have not been characterized in detail. Here, we performed global gene expression profiling of the brain and testis tissues of three positive transgenic male offspring (genotype: hDNMT3A+/-), three negative transgenic male offspring (genotype: hDNMT3A-/-) and three wild-type male rats (genotype: hDNMT3A-/-) to identify the impact of hDNMT3A overexpression on the tissue transcriptome and to investigate whether the impact could be intergenerationally inherited without the transmission of the initial endogenic trigger (). We subsequently characterized the DNA methylation patterns of the derivative differentially expressed genes to analyse whether their expression variations were affected by DNA methylation changes on their promoters, as DNMT3A acted as a de novo methyltransferase. In this study, both positive transgenic offspring and negative transgenic offspring were obtained from the same batch of litter, which derived from the same hDNMT3A+/- farther and the wild-type mother ()). Although negative transgenic offspring and wild-type rats had the same genotype, hDNMT3A-/-, negative transgenic offspring might have different epigenome with wild-type rats if DNA methylation variations and gene expression variations could be intergenerationally inherited (i.e. epigenetic inheritance). Thus, positive transgenic offspring, negative transgenic offspring and wild-type rats were represented as hDNMT3A+/-, hDNMT3A-/- and WT, respectively.
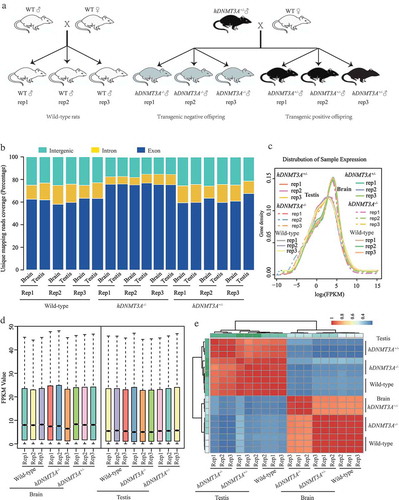
Figure 1. RNA-seq analysis of tissues from hDNMT3A+/-, hDNMT3A-/- and WT rats. a. The rat lines used in this study. b. Unique mapping reads coverage. c. Distribution analysis of gene expression density in different samples. d. Distribution analysis of the FPKM values in different samples. e. Heatmap of the samples’ correlation.
Results
RNA-seq analysis of tissues from hDNMT3A+/-, hDNMT3A-/- and WT rats
To investigate the transcriptional responses of rats to the hDNMT3A transgene, RNA from the brain and testis tissues of hDNMT3A+/-, hDNMT3A-/- and WT rats was extracted to construct cDNA libraries for RNA-seq analysis on an Illumina HiSeq 4000 platform (Illumina, CA). After adaptor trimming and removal of low-quality reads, we obtained approximately 46.5 million clean reads per sample on average (Supplemental Table 1). The data showed that over 92% of the clean reads could be mapped to the reference genome and the multimapped reads accounted for only ~6% of the reads (Supplemental Table 1). Among the unique mapped reads, approximately 60%, 15% and 25% were distributed on exons, introns and intergenic regions, respectively ()). In addition, more than 16,500 genes could be mapped for each sample (Supplemental Table 1). These results suggested that our RNA-seq data were sufficient for subsequent gene expression analysis.
Our results revealed that the brain and testis tissues exhibited different expression patterns (), but there were no significant differences in gene expression density and FPKM value among three rat lines in either the brain or testis (,)). We then performed correlation analysis, and the results revealed that the brain and testis samples clustered into two distinct groups, indicating that the differences in gene expression patterns between tissues were greater than those between rat lines ()). However, in both subgroups, tissues from the same rat lines clustered together, suggesting that the hDNMT3A transgene affected the transcriptomes of the rat brain and testis ()). Thus, although the differences between rat lines were smaller than those between tissues, hDNMT3A expression did influence the gene expression patterns of the two tissues.
Expression patterns of hDNMT3A and rat-derived DNMT3A (rDNMT3A)
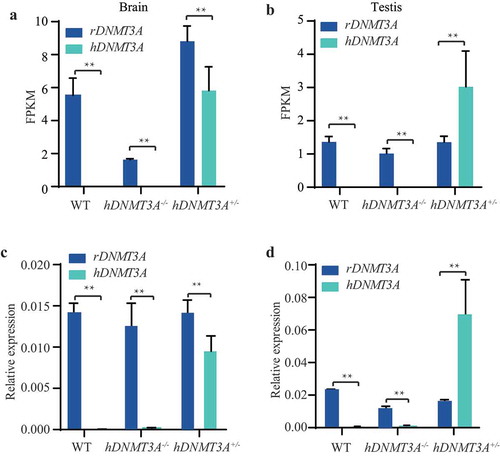
To validate whether hDNMT3A (NCBI-ID:1788) was specifically expressed in the transgenic rats, we analysed the expression patterns of hDNMT3A and rDNMT3A (NCBI-ID:444,984) in the brain and testis of hDNMT3A+/-, hDNMT3A-/- and WT rats. The results showed that hDNMT3A was specifically expressed in hDNMT3A+/- tissues (, )). In the brain of hDNMT3A+/- rats, hDNMT3A exhibited the same expression pattern as rDNMT3A, whereas in the testis, the hDNMT3A expression level was approximately 2-fold higher than the rDNMT3A expression level (, )). The same results were obtained in the qPCR validation experiments (, )). Although the same expression levels of rDNMT3A were observed between transgenic offspring and WT rats in both the brain and testis, the additional expression of hDNMT3A in hDNMT3A+/- rats implied that DNMT3A was overexpressed in hDNMT3A+/- rat tissues.
Figure 2. Expression patterns of hDNMT3A and rDNMT3A in all rat lines. The expression patterns of hDNMT3A and rDNMT3A in the brain (a) and testis (b) detected by mRNA-seq. The expression patterns of hDNMT3A and rDNMT3A in the brain (c) and testis (d) validated by qPCR.
Additional expression of hDNMT3A induced gene expression variations in the brain and testis
To identify the impact of the additional expression of hDNMT3A on tissue transcriptome, we analysed the differences in gene expression patterns between hDNMT3A+/- and WT rats in both the brain and testis. The results showed that there were 1,041 and 1,373 differentially expressed genes in the brain and testis, respectively (, )). Among these differentially expressed genes between hDNMT3A+/- and WT rats, downregulated genes accounted for 46.39% in the testis and approximately 48.22% in the brain (, )).
Figure 3. Differentially expressed genes between transgenic offspring and WT rats.
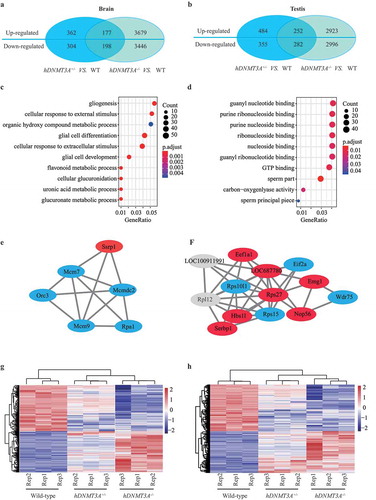
To investigate the functions of the differentially expressed genes, we conducted a Gene Ontology (GO) analysis and found that they were involved in the regulation of glial cell differentiation, cellular response to external stimulus and cellular glucuronidation in the brain ()). In the testis, they had nucleoside binding and carbon−oxygen lyase activity and participated in DNA recombination, translation and peptide metabolic processes ()). We then performed protein-protein interaction (PPI) analysis to study the functional relationship of these differentially expressed genes. Based on the 1,041 differentially expressed genes in the brain, the PPI enrichment p-value was 1.5 × 10−9. The sub-cluster genes participated in ribosome biogenesis and assembly, regulation of translation (Supplemental Figure 1a and 1b), DNA replication and DNA repair ()), Supplemental Figure 1c and 1d). In the testis, based on the 1,373 differentially expressed genes, the PPI enrichment p-value was 1.0 × 10−16. The sub-cluster genes participated in rRNA and ncRNA processing (), Supplemental Figure 1 e-h). Differentially expressed genes induced by additional expression of hDNMT3A were involved in different functions between the two tissues, suggesting that its overexpression had different effects on the two tissues.
In addition, between the WT and hDNMT3A+/- rats, 94 differentially expressed genes were common to both the brain and testis, including 52 upregulated genes and 42 downregulated genes (Supplemental Figure 2 a-c). The similar tendencies in both tissues suggested that the expression patterns of these genes were very likely affected by the additional expression of hDNMT3A. Further analysis revealed that most of these genes, including Dnmt3a, Sox9 and so on, had binding and catalytic activity and participated in biological regulation, cellular and metabolic processes.
Intergenerational inheritance of the transgene-induced gene expression variations
To investigate whether these gene expression variations could be inherited to offspring without the transmission of the transgene, we analysed the transcriptome of the brain and testis tissues of the transgenic negative offspring. The results showed that, compared with WT, hDNMT3A+/- and hDNMT3A-/- rats had 549 common differentially expressed genes in the brain tissues (Supplemental Figure 2d) and had 722 common differentially expressed genes in the testis tissues (Supplemental Figure 2e). Among which, 430 genes and 390 genes also differentially expressed between hDNMT3A+/- and hDNMT3A-/-, respectively (Supplemental Figure 2d and 2e). Whereas, some of these common differentially expressed genes might have different tendencies in different comparison combinations, i.e. upregulated in one comparison combination but downregulated in another comparison combination. Further analysis revealed that, in the brain, there were 375 differentially expressed genes that had the same tendencies between the comparison of hDNMT3A+/- and WT rats and between the comparison of hDNMT3A-/- and WT rats, including 177 common upregulated genes and 198 common downregulated genes, accounting for 36.02% of total differentially expressed genes which could be induced by the additional expression of hDNMT3A ()). In the testis, the number was 534, including 252 common upregulated genes and 282 common downregulated genes, accounting for 38.89% of total differentially expressed genes ()). Although some of these common genes also differentially expressed between hDNMT3A+/- and hDNMT3A-/- rats, the variations were less than that between transgenic offspring (both hDNMT3A+/- and hDNMT3A-/-) and WT rats in both tissues (, )). Thus, we considered that these common differentially expressed genes between transgenic offspring and WT rats could be intergenerationally inherited to hDNMT3A-/- rats without the transmission of the transgene.
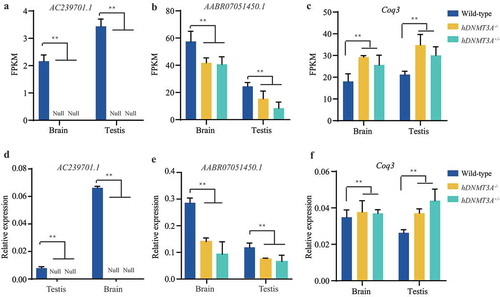
To validate the reliability of the sequencing data, we performed qPCR to detect the expression patterns of ten differentially expressed genes, including AC239701.1, AABR07051450.1, Ccndbp1, Coq3, Cyren, Acox3, Cand2, Galns, Pitpnm1 and Retsat ( and supplemental Figure 3). The results from the two experiments were identical, suggesting that the sequencing data were stable and reliable for the inheritance analysis. Meanwhile, the changed expression patterns of AC239701.1, AABR07051450.1, Ccndbp1 and Coq3 in both the brain and testis tissues and Cyren, Acox3, Cand2, Galns and Pitpnm1 in the testis could be also intergenerationally inherited to transgenic negative offspring ( and supplemental Figure 3). These observations indicated the intergenerational inheritance of gene expression variations, which were induced by the additional expression of hDNMT3A, without the transmission of the initial trigger.
Figure 4. Using qPCR to validate differentially expressed genes between rat lines.
Intergenerational inheritance of the DNA methylation variations which associated with the differentially expressed genes
Epigenetic mechanisms have been reported to play important roles in the transmission of parental life experiences to offspring and could mediate long-lasting changes in gene expression, even when the initial triggering signal has disappeared. In addition, gene expression levels can be modulated by the DNA methylation status, and hDNMT3A represents a de novo DNA methyltransferase. We then detected the DNA methylation statuses of AC239701.1, AABR07051450.1 and Coq3 in both tissues of all three rat lines by using the bisulphite sequencing PCR (BSP) method to validate whether the DNA methylation changes were associated with their expression variations and mediated the intergenerational inheritance. The CpG islands that we detected in AC239701.1 and AABR07051450.1 were located in their promoters while the CpG island that we detected in Coq3 was located in the first exon. The results showed that the DNA methylation levels of both the AC239701.1 and AABR07051450.1 promoters were significantly higher in hDNMT3A+/- and hDNMT3A-/- rats than in WT rats in both tissues ()). Increased promoter DNA methylation levels integrated with the down-regulated expression of these two genes suggested that their expression variations were negatively correlated with the promoter DNA methylation level. Nevertheless, in Coq3 exon, although DNA methylation levels in all three rat lines were low than 10%, the methylation status in hDNMT3A+/- and hDNMT3A-/- rats were higher than in WT rats, indicating positive correlation between exon methylation and gene expression, which was consistant with the other’s study [Citation18] (Supplemental Figure 4). Furthermore, similar DNA methylation patterns of these genes in hDNMT3A-/- rats to that in hDNMT3A+/- rats indicated that the DNA methylation variations could be intergenerationally inherited without the transmission of the transgene (, Supplemental Figure 4). All these observations suggested that the heritable DNA methylation variations would be the mechanism underlying intergenerational inheritance of gene expression variations.
Figure 5. Intergenerational inheritance of the transgene-induced DNA methylation variations.
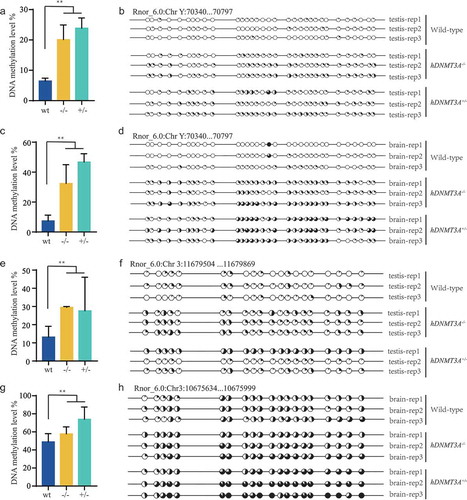
In addition, we also observed that, although might have different tendencies, there were 589 and 673 differentially expressed genes between hDNMT3A+/- genotype and hDNMT3A-/- genotype rats (including WT and negative transgenic rats) in the brain and testis, respectively (Supplemental Figure 2d and 2e). The expression patterns of these genes would be affected by DNA methylation variations which were mediated by the overexpression of the de novo methyltransferase, DNMT3A, in hDNMT3A+/- rats. To validate this speculation, we identified the promoter DNA methylation pattern of Dkc1, a gene that differentially expressed between hDNMT3A+/- and hDNMT3A-/- genotype rats in both tissues (Supplemental Figure 5a and 5b). The results showed that hDNMT3A+/- genotype rats had higher DNA methylation levels than hDNMT3A-/- genotype rats in Dkc1 promoter of both brain and testis tissues (Supplemental Figure 5c and 5d). Although differentially expressed between two genotypes, Dkc1 had high expression levels but low DNA methylation levels in both tissues of all three rat lines (Supplemental Figure 5e and 5f). These results revealed that there were some genes with down-regulated expressions but increased DNA methylation levels only in hDNMT3A+/- genotype, suggesting that the overexpression of DNMT3A in hDNMT3A+/- rats induced the de novo methylation and subsequently mediated the downregulation of the genes.
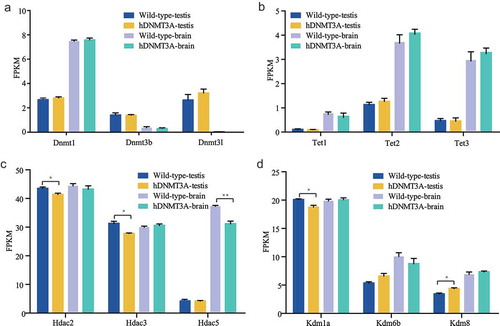
Impact of additional expression of hDNMT3A on other epigenetic modifiers
The expression patterns of genes are influenced by transcription factors and chromatin structure, and the latter is modulated by epigenetic modifications, including DNA methylation and histone covalent modifications. To determine whether the additional expression of hDNMT3A affected other epigenetic modifiers, we analysed the expression levels of modifiers, such as other DNMTs (such as Dnmt1, Dnmt3b and Dnmt3l), Tet family members associated with demethylation (such as Tet1, Tet2 and Tet3), histone deacetylases (such as Hdac1–Hdac11), lysine demethylases (such as Kdm1a–Kdm8), and euchromatic histone lysine methyltransferases (such as Ehmt1 and Ehmt2). The results revealed that the other DNMTs did not differ in expression between the WT and hDNMT3A+/- rats in either the brain or testis ()). Although the expression levels of Tet2 and Tet3 were higher in the brains of hDNMT3A+/- rats than in those of WT rats, the differences were not statistically significant ()). Interestingly, Hdac2 and Hdac3 were significantly downregulated in the testes of hDNMT3A+/- rats, while Hdac5 was significantly downregulated in the brain ()). In addition, Kdm1a was downregulated but Kdm6b and Kdm8 were upregulated in the testes of transgenic rats ()). Epigenetic modifiers, including DNA and histone modifiers, interplay with each other in regulating gene expression. These results suggested that the additional expression of hDNMT3A affected the expression patterns of the other epigenetic modifiers. Their expression variations might play antagonistic or compensatory roles in response to the additional expression of hDNMT3A in regulating gene expression patterns.
Figure 6. Impact of additional expression of hDNMT3A on other epigenetic modifiers.
Discussion
DNA methylation plays critical roles in transcriptional regulation and the timely establishment of maternal and paternal imprints during embryonic development and gametogenesis [Citation19]. DNMTs, including Dnmt1, Dnmt3a, Dnmt3b, Dnmt3c and Dnmt3l, are responsible for establishing and maintaining the DNA methylation status. Previous studies have revealed that deficiency in Dnmt3a or Dnmt3b leads to embryonic lethality in mice and that Dnmt3a overexpression is related to tumorigenesis in humans [Citation7,Citation20,Citation21]. In this study, we used our previously constructed humanized rat model that expressed hDNMT3A to investigate the impact of additional DNMT3A expression on brain and testis transcriptome profiles and the DNA methylation patterns of differentially expressed genes. Meanwhile, we investigated whether these induced gene expression and DNA methylation variations could be intergenerationally inherited without the transmission of the initial endogenic trigger. Our results showed that hDNMT3A was overexpressed in the testis, which is consistent with the results of our previous study; however, the hDNMT3A expression levels in the brain were lower than those in our previous study, which could be due to individual differences [Citation17].
We found that the additional expression of hDNMT3A made a notable impact on the transcriptomes of both the testis and brain and that most of the affected genes were different between the two tissues; therefore, the affected genes had different molecular functions and participated in different biological processes in the two tissues. These observations suggested that hDNMT3A might have different functions in the testis and brain [Citation22,Citation23]. In addition, further study revealed that the expression patterns of some of the differentially expressed genes between WT and hDNMT3A+/- rats were negatively correlated with the DNA methylation status of their promoters, which would have been induced by the additional expression of hDNMT3A ( and ), and these findings are consistent with the action of DNMT3A as a de novo DNMT and with a previous study in which DNA methyltransferase was shown to affect gene expression patterns through modulating DNA methylation status on their promoters [Citation24]. Histone modifiers, such as Hdacs and Kdms, as well as proteins related to demethylation, such as the Tet family proteins, also interact with DNMTs in regulating chromatin structure, accessibility and gene expression [Citation24–Citation26]. In previous studies, the abnormal expression of DNMTs and histone modifiers has been reported to lead to diseases or even lethality in mammals [Citation16,Citation27,Citation28]; however, in this study, these epigenetic modifiers with variations in expression levels might play antagonistic or compensatory roles in responding to hDNMT3A overexpression and result in steady and balanced tissue functions. Such roles may explain why we did not observe significant behavioural and reproductive phenotypes in hDNMT3A+/- rats despite the thousands of genes in the brain and testis that were affected by additional expression of hDNMT3A. Notably, DNMT3A expression was significantly associated with human gastric and hepatocellular carcinoma prognoses [Citation12,Citation13]; therefore, additional research is needed to confirm whether these transgenic rats are sensitive to tumours and cancer [Citation17].
The most important discovery in this study was that the overexpression of hDNMT3A induced intergenerational inheritance of DNA methylation and gene expression variations in rat brains and testes without the transmission of the transgene. In a previous study, conditional overexpression of hKDM1A in the testes of male mice reduced H3K4me2 in mature sperm, resulting in severely impaired development and offspring survival, and the phenotype and altered histone modification pattern could be transmitted through at least four generations without the transmission of hKDM1A [Citation16]. Our previous study illustrated that overexpression of hDNMT3A in rat testes can induce intergenerational inheritance of DNA methylation variations in sperm to negative-transgenic offspring [Citation17]. Here, we further demonstrated that transcriptome and methylome variations induced by the aberrant expression of endogenous epigenetic modifiers could not only be intergenerationally inherited to germ cells of the offspring, such as the sperm, but also could be succeeded in the corresponding tissues of the offspring. These intergenerationally inherited gene expression and DNA methylation variations were considered actively induced by endogenous factors, which is different from intergenerationally or transgenerationally inherited gene expression and DNA methylation variations that are passively induced by environmental factors [Citation17,Citation29]. Further study is necessary to determine the number of generations without hDNMT3A that can stably inherit differential gene expression and promoter DNA methylation changes initially caused by the additional expression of hDNMT3A.
Conclusions
Using our previously established humanized rat model and through mRNA-sequencing and bisulphite sequencing PCR, we identified the impact of hDNMT3A overexpression on tissue transcriptome and methylome and subsequently investigated whether the variations in gene expression and DNA methylation could be inherited intergenerationally. Our results revealed that the additional expression of hDNMT3A could induce notable gene expression changes in the rat testis and brain, and these gene expression variations were negatively correlated with DNA methylation variations in their promoters. More importantly, these gene expression and DNA methylation variations could be intergenerationally inherited to offspring tissues without the transmission of the initial endogenic trigger, i.e., the hDNMT3A transgene. Compared with inheritable variations that were passively induced by environmental factors, these variations were actively induced by endogenous epigenetic modifiers. This study provided evidence for the epigenetic inheritance of endogenous factors that actively induce gene expression and DNA methylation variations, although more studies are needed to determine the number of generations that can stably inherit differential gene expression and promoter DNA methylation changes initially caused by hDNMT3A overexpression.
Materials and methods
Animals
All studies involving animals were conducted under the approval of the Institutional Animal Care Committee of Shanghai Institute of Biochemistry and Cell Biology. The hDNMT3A transgenic rats were obtained from our previous study [Citation17]. In brief, the bacterial artificial chromosome (RP11-159D24, NCBI-ID:223335) containing human-derived DNMT3A was injected into rat zygotes (Rattus norvegicus, Sprague-Dawley, SD). The positive transgenic rats (F0) were identified by PCR with the corresponding primers: PCR-hDNMT3A-F: 5'-GCCTCAGAGCTATTACCCAATGG-3' and PCR-hDNMT3A-R: 5'-GGAGAGTAAGCAGGCCAGGTAGAG-3'. After two generations of the species preservative propagation, one of the positive transgenic male rats (, ♂, F2) was mated with a wild-type female rat (WT, ♀), their offspring (F3) inhDNMT3A+/-cluding positive transgenic rats (hDNMT3A+/-) and negative transgenic rats (hDNMT3A-/-, )). Although wild-type rats and negative transgenic offspring had the same genotype, they might have different epigenome-type. Therefore, in this study, we used hDNMT3A+/- and hDNMT3A-/- to represent positive and negative transgenic offspring, respectively. Wild-type rats were represented by WT. Finally, WT male rats, hDNMT3A+/- male offspring (F3) and hDNMT3A-/- male offspring (F3) were used in this study.
RNA extraction and mRNA-seq
Total RNA was extracted from the hippocampus and testis of WT, hDNMT3A+/- and hDNMT3A-/- rats using RNeasy Plus Universal Kits (Qiagen, Germany) according to the manufacturer’s instructions. The concentration and quality of the total RNA were determined with a NanoDrop 2000 (Thermo Fisher Scientific, U.S.) and a 2100 Bioanalyzer (Agilent, U.S.). A total of 5 μg of high-quality RNA was used to construct each sequencing library.
The mRNA-seq libraries were prepared with a TruSeq Stranded Total RNA Preparation Kit (Illumina, CA) according to the manufacturer’s instructions. cDNA synthesis, end repair, A-base addition and ligation of the Illumina-indexed adaptors were performed according to the Illumina protocol. The paired-end libraries were sequenced with 2 × 150-bp (PE150) reads on an Illumina HiSeq 4000 instrument. Three biological replicates were performed for each genotype.
Mapping and processing of mRNA-seq reads
The raw data were trimmed and subjected to quality control. Then, the clean reads were separately aligned to the reference genome. The expression level of each transcript was calculated using the fragments per kilobase of exon per million mapped reads (FPKM) method. Differentially expressed genes between two samples were identified based on a cut-off P value for per-gene variance less than 0.05 with conservative Bonferroni correction. Protein and Protein interaction analysis was performed in the web: https://string-db.org/.
qPCR to validate the differentially expressed genes
Total RNA (1 μg) was used to synthesize cDNA with an EasyScript One-Step gDNA Removal and cDNA Synthesis SuperMix kit (TransGen, China) according to the manufacturer’s instructions. The qPCR volume contained 0.5 μl of synthesized cDNA, 10 μl of 2x TransStart Top Green qPCR SuperMix (TransGen, China), 1.6 μl of forward primer (2.5 μM), 1.6 μl of reverse primer (2.5 μM), 0.4 μl of Passive Reference Dye II (50x) and ddH2O to a final volume of 20 μl. The PCR procedure included an initial step of 95°C for 30 s; 40 cycles of 95°C for 5 s and 60°C for 34 s; and a dissociation curve step on an ABI Prism 7500 Real-Time Thermal Cycler (Applied Biosystems, Life Science, U.S.). Three biological replicates and three technical replicates were used. The results were normalized by those for the rat housekeeping gene rGapdh. The primers used in this study were listed in Supplemental Table 2.
Bisulphite sequencing PCR (BSP) to identify promoter DNA methylation levels
We used BSP, a gold standard method for measuring DNA methylation patterns, to identify the DNA methylation statuses of the differentially expressed gene promoters. After bisulphite conversion, methylated cytosine remained unchanged while unmethylated cytosine was converted to uracil. After amplification by PCR, the unmethylated cytosine changed to thymine. The DNA methylation status of a targeted cytosine could therefore be determined by comparing the PCR product sequence with the reference sequence. An EpiMark Bisulphite Conversion Kit (NEB, U.S.) was used to convert 1 μg of genomic DNA from each of the two tissues according to the manufacturer’s instructions. After being dissolved in 30 μl of ddH2O, the converted product was subjected to PCR. The reaction mixture contained 10 μl of 5x EpiMark Hot Start Taq Reaction Buffer (NEB, U.S.), 1 μl of dNTPs (10 mM, Takara, Japan), 1 μl of forward primer (10 μM), 1 μl of reverse primer (10 μM), 0.25 μl of EpiMark Hot Start Taq DNA Polymerase (NEB, U.S.), 6 μl of converted genomic DNA, and ddH2O to a final volume of 50 μl. The PCR procedure included an initial step of 95°C for 30 s; 45 cycles of 95°C for 30 s, 45°C for 30 s and 68°C for 30 s; a final step of 68°C for 5 min; and a hold at 4°C. The PCR product was purified with an EasyPure Quick Gel Extraction Kit (TransGen, China) and subsequently cloned into a pEasy-T1 vector (TransGen, China). Then, individual clones for each sample were sequenced to analyse the DNA methylation pattern of the targeted cytosine. The calculate process was conducted on the QUMA website: http://quma.cdb.riken.jp/. The primers used in this study were listed in Supplemental Table 2.
Author Contributions
FS and XZ designed the work, ZL wrote the manuscript, YM constructed the hDNMT3A transgenic rats, ZL and YZ performed the RNA-seq and BSP assays, GW and HW performed the qPCR assay to analyzed the expression patterns of transgene and differentially expressed genes, YD performed tissue DNA extraction, and XZ and SC analyzed the qPCR data.
Availability of data and materials
All datasets generated in this work have been deposited in the Sequence Read Archive (SRA) database under the accession number GSE128777.
Supplemental Material
Download MS Word (2.3 MB)Acknowledgments
We would like to thank Dr. Yu Lin for providing a discussion and critical reading of the manuscript. We thank Shanghai Key Laboratory of Reproductive Medicine, School of Medicine, Shanghai Jiao Tong University for the animal care and equipment support.
Disclosure statement
The authors declare that they have no competing interests.
Supplementary material
Supplemental data for this article can be accessed here
Additional information
Funding
Related Research Data
References
- Li E, Zhang Y. DNA methylation in mammals. Cold Spring Harb Perspect Biol. 2014;6:a019133.
- Jeong M, Sun D, Luo M, et al. Large conserved domains of low DNA methylation maintained by Dnmt3a. Nat Genet. 2014;46(1):17-+.
- Auclair G, Weber M. Mechanisms of DNA methylation and demethylation in mammals. Biochimie. 2012;94(11):2202–2211.
- Law JA, Jacobsen SE. Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat Rev Genet. 2010;11(3):204–220.
- Barau J, Teissandier A, Zamudio N, et al. The DNA methyltransferase DNMT3C protects male germ cells from transposon activity. Science. 2016;354(6314):909–912.
- Goll MG, Kirpekar F, Maggert KA, et al. Methylation of tRNA Asp by the DNA Methyltransferase Homolog Dnmt2. Science. 2006;311(5759):395–398.
- Okano M, Bell DW, Haber DA, et al. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell. 1999;99(3):247–257.
- Spencer DH, Russler-Germain DA, Ketkar S, et al. CpG Island Hypermethylation Mediated by DNMT3A Is a Consequence of AML Progression. Cell. 2017;168(5):801–816. e13.
- Qu Y, Mu G, Wu Y, et al. Overexpression of DNA Methyltransferases 1, 3a, and 3b significantly correlates with retinoblastoma tumorigenesis. Am J Clin Pathol. 2010;134(5):826–834.
- Rahman MM, Qian ZR, Lu Wang E, et al. DNA methyltransferases 1, 3a, and 3b overexpression and clinical significance in gastroenteropancreatic neuroendocrine tumors. Hum Pathol. 2010;41(8):1069–1078.
- Siddiqui NN, Ul Haq A, Siddiqui OA, et al. DNA methyltransferase 1, 3a, and 3b expression in hepatitis C associated human hepatocellular carcinoma and their clinicopathological association. Tumor Biol. 2016;37(8):10487–10497.
- Cao X-, Ma H, Shang Y, et al. DNA methyltransferase3a expression is an independent poor prognostic indicator in gastric cancer. World J Gastroenterol. 2014;20(25):8201–8208.
- Oh BK, Kim H, Park H-J, et al. DNA methyltransferase expression and DNA methylation in human hepatocellular carcinoma and their clinicopathological correlation. Int J Mol Med. 2007;20(1):65–73.
- Kadriu B, Guidotti A, Chen Y, et al. DNA methyltransferases1 (DNMT1) and 3a (DNMT3a) colocalize with GAD67-positive neurons in the GAD67-GFP mouse brain. J Comp Neurol. 2012;520(9):1951–1964.
- Kaneda M, Okano M, Hata K, et al. Essential role for de novo DNA methyltransferase Dnmt3a in paternal and maternal imprinting. Nature. 2004;429(6994):900–903.
- Siklenka K, Erkek S, Godmann M, et al. Disruption of histone methylation in developing sperm impairs offspring health transgenerationally. Science. 2015;350(6261):aab2006.
- Zheng X, Li Z, Wang G, et al. Overexpression of human-derived DNMT3A induced intergenerational inheritance of active DNA methylation changes in rat sperm. Front Genet. 2017;8:207.
- Yang X, Han H, De Carvalho D, et al. Gene body methylation can alter gene expression and is a therapeutic target in cancer. Cancer Cell. 2014;26(4):577–590.
- Uysal F, Ozturk S, Akkoyunlu G. DNMT1, DNMT3A and DNMT3B proteins are differently expressed in mouse oocytes and early embryos. J Mol Histol. 2017;48(5–6):417–426.
- Leonard S, Pereira M, Fox R, et al. Over-expression of DNMT3A predicts the risk of recurrent vulvar squamous cell carcinomas. Gynecol Oncol. 2016;143(2):414–420.
- Chen BF, Chan WY. The de novo DNA methyltransferase DNMT3A in development and cancer. Epigenetics. 2014;9(5):669–677.
- Galetzka D, Tralau T, Stein R, et al. Expression of DNMT3A transcripts and nucleolar localization of DNMT3A protein in human testicular and fibroblast cells suggest a role for de novo DNA methylation in nucleolar inactivation. J Cell Biochem. 2006;98(4):885–894.
- Feng J, Chang H, Li E, et al. Dynamic expression of de novo DNA methyltransferases Dnmt3a and Dnmt3b in the central nervous system. J Neurosci Res. 2005;79(6):734–746.
- Gu T, Lin X, Cullen SM, et al. DNMT3A and TET1 cooperate to regulate promoter epigenetic landscapes in mouse embryonic stem cells. Genome Biol. 2018;19(1):88.
- Zhang R, Erler J, Langowski J. Histone acetylation regulates chromatin accessibility: role of H4K16 in inter-nucleosome interaction. Biophys J. 2017;112(3):450–459.
- Bustos MA, Salomon MP, Nelson N, et al. Genome-wide chromatin accessibility, DNA methylation and gene expression analysis of histone deacetylase inhibition in triple-negative breast cancer. Genom Data. 2017;12:14–16.
- Bergman Y, Cedar H. DNA methylation dynamics in health and disease. Nat Struct Mol Biol. 2013;20(3):274–281.
- Fu X, SONG X, LI Y, et al. Hepatitis B virus X protein upregulates DNA methyltransferase 3A/3B and enhances SOCS-1CpG island methylation. Mol Med Rep. 2016;13(1):301–308.
- Gapp K, von Ziegler L, Tweedie-Cullen RY, et al. Early life epigenetic programming and transmission of stress-induced traits in mammals: how and when can environmental factors influence traits and their transgenerational inheritance? Bioessays. 2014;36(5):491–502.