
ABSTRACT
The current decline in dairy cattle fertility has resulted in significant financial losses for dairy farmers. In the past, most efforts to improve dairy cattle fertility have been focused on either management or genetics, while epigenetics have received less attention. In this study, 12 bulls were selected from a provided 100 bull list and studied (High daughter fertility = 6, Low daughter fertility = 6) for Enzymatic methylation sequencing in the Illumina HiSeq platform according to the Canadian daughter fertility index (DFI), sires with high and low daughter fertility have average DFI of 92 and 112.6, respectively. And the bull list provided shows a mean DFI of 103.4. 252 CpGs with methylation differences greater than 20% (q < 0.01) were identified, as well as the top 10 promising DMRs with a 15% methylation difference (q < 1.1e−26). Interestingly, the DMCs and DMRs were found to be distributed more on the X chromosome than on the autosome, and they were covered by gene clusters linked to germ cell formation and development. In conclusion, these findings could enhance our ability to make informed decisions when deciding on superior bulls and advance our understanding of paternal epigenetic inheritance.
Introduction
Over the past five decades, as a result of intensive selection pressure, both dairy and beef cattle have shown a decline in fertility efficiency [Citation1,Citation2] resulting in huge economic profitability losses to dairy farmers. Advances in technology such as NGS (Next-generation sequencing) and GWAS (Genome-wide association analysis) have enabled many of the Single-nucleotide Polymorphism (SNPs) and Quantitative trait locus (QTLs) associated with fertility to be identified [Citation3] and allowed us a systematic understanding of the genetic basis of this low heritability trait [Citation4]. For instance, four SNPs (rs110068451, rs109983109, rs110543856, and rs29027634) that were validated for more than one cow trait increased confidence that female fertility is associated with these SNPs [Citation3]. Given the fact that fertility phenotype is determined by genotype, environmental factors, and epigenotype [Citation5], developing epigenomic strategies for improving dairy cattle fertility should be based upon an integrated approach for the benefit of dairy farmers and cow welfare.
Recently, there has been some evidence that abnormal DNA methylation in sperm is associated with male fertility [Citation6], embryo quality [Citation7,Citation8], and the susceptibility of offspring to disorders [Citation9–11] in humans and animals including bovine. It is known that the information beyond the DNA sequence can be passed on to future generations by gametes [Citation11] given the incomplete erasure of the DNA methylation during the early gametogenesis and embryogenesis [Citation12,Citation13]. As an example of the transmitted epigenome, our previous study indicated that metabolically stressed cows could influence the DNA methylation of the offspring’s blood cells [Citation14], and the altered DNA methylation in F1 blood was associated with the metabolic pathways. In our recently published work [Citation15], we successfully identified the sperm-derived bull fertility-associated DNA methylation markers, in the current study, we applied the enzyme-based whole genome methylation sequencing to screen the daughter fertility-associated methylation markers, in order to integrate into the existing approach for dairy cattle fertility predicting/improving.
As a result, this study hypothesizes that some DNA methylation characteristics in bovine sperm can resist epigenetic reprogramming, thereby passing on to the F1 female offspring, and associated with the F1 phenotype. Using the daughter fertility index [Citation16,Citation17], which consists of several fertility traits, we determined the bulls with high and less daughter fertility, sequenced and analysed their sperm methylation characteristics, and expected to find the epi-markers correlated with daughter fertility. As a result of these methylation characteristics, we will be able to make better decisions regarding the bull selection and dairy breed fertility improvements, references will also be provided for future fertility-related studies and epigenetic knowledge will be available on the intergenerational transmission of epigenetic traits in domestic animals.
Results
Differentially methylated cytosines (DMCs) associated with daughter fertility
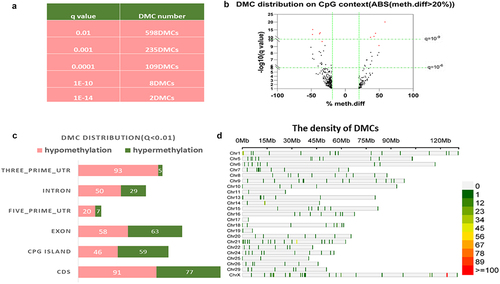
To identify the overall differentially methylated patterns in bovine sperm DNA that are associated with daughter fertility, from a pre-filtered 100 bulls, six bulls with high daughter fertility (DFI >111) and six bulls with low daughter fertility (DFI >94) were chosen (Supplementary file 1) for whole-genome methylation comparisons The read coverage per base of less than 10× was filtered out. Data were deposited at the National Center of Biotechnology Information (NCBI) under accession number GSE211926. According to Supplementary File 2, each sample exhibited an enzymatic conversion rate of 94.37% to 99%. In total 17,730 CGs were identified. By adjusting the q-value cut-off, the number of significant CGs varied. When q < 0.01 (), 598 CGs were identified. Most of these CGs were distributed in Coding DNA Sequence (CDS) regions (), exons, and Cytosine-phosphate-Guanine (CpG) islands. Supplementary file 1 shows that we identified 252 DMCs with a methylation difference greater than 20% (q < 0.05), among which, a total of 64 DMCs were hypermethylated, while 188 DMCs were hypomethylated. As shown in the volcano plot in , the top 10 most promising DMCs had at least 35% methylation difference (q < 10−9) were identified and coloured red. Interestingly, the density of single-site DNA methylation on each chromosome varied (), it is interesting to note that the majority (28.5%) of the significant DMCs are located on the X chromosome (). Specifically, paternal single-site DNA methylation was associated with their daughters’ fertility, with the variations mostly occurring on chromosome X.
Figure 1. Identification of daughter fertility – associated differentially methylated cytosines in bovine sperm. a. Table of the filtering DMCs via q-value. b. Volcano plot of the daughter fertility correlated CpGs that identified by 20% methylation difference. The horizontal green lines represent q-values that equal 10−6 (lower) and 10−10 (upper). The vertical green lines represent a − 20% methylation difference (left) and a 20% methylation difference (right). The red dots represent the top 10 promising DMCs correlated with daughter fertility. c. The bar chart shows the distribution of the CGs in different genomic elements (UTR: untranslated region, CDS: coding sequence). d. The density of the CGs (q < 0.01). The colour represents the number of DMC.
Differentially methylated regions (DMRs) associated with daughter fertility
In order to determine the methylation regions that may be associated with daughter fertility phenotypes, we further extracted DMRs between high and low daughter fertility groups. By using the methylkit package, we found 7,291 differentially methylated bins with a sliding bin size of 500 bp and a step of 100 bp, which equals 1,629 DMRs (Supplementary Figure S1). shows that the majority of bins are located on CpG islands (q < 0.01), with 1371 hypomethylated and 347 hypermethylated bins, and fewer bins were located in untranslated regions, namely, 149 in 3’Untranslated region (UTR) and 192 in 5’UTR.In agreement with the single-site methylation results, most bins were hypomethylated.
Figure 2. The distribution of the candidate differentially methylated regions correlated with daughter fertility. a. The bar chart depicts the genomic distribution of the bins correlated with daughter fertility, the yellow colour indicates hypomethylation and the purple colour represents hypermethylation. b. The chromosome location of the bins correlated with daughter fertility (orange indicates hypermethylation; brown indicates hypomethylation) (CpG: CpG island, CDS: coding DNA sequence, UTR: untranslated region).
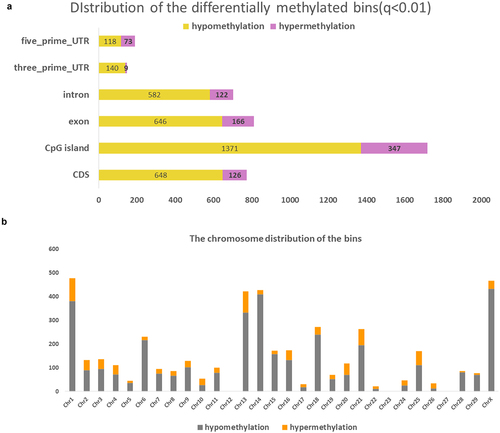
Besides, in line with the CG distribution, most bins associated with daughter fertility were located on chromosome X (). When a methylation difference higher than 15% and a stringent q-value of 1.1e−26 were applied, the top 10 DMRs were identified and some of the DMRs also covered the promising DMCs (Supplementary Table S1). For example, two DMCs were found in DMR6, located at ChrX: 123940140 and ChrX: 123940191, with methylation differences of −35.251% and −25.42%, respectively. ChrX:124021747 was the DMC covered by DMR8 with a methylation difference of −34.806%. In summary, a phenotype related to daughter fertility was found in the paternal sperm differentially methylated regions, which were located mostly on the X chromosome.
Gene ontology (GO) and gene function cluster analysis
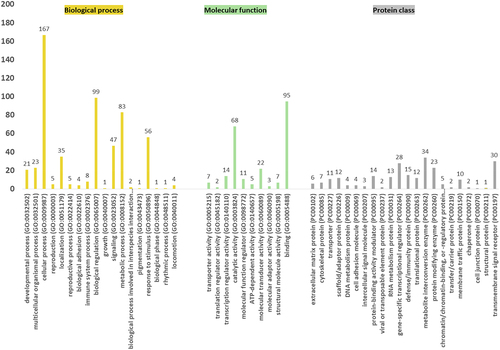
In order to better understand the potential function of the identified DMRs, we annotated the screened DMRs using the annotate peak function of the ChIPseeker R package. The biological functions of the 355 genes encompassed at least one DMR (q < 0.05) were examined with Panther using GO slim analysis on the genes. According to , the most enriched GO terms under the biological process category were cellular process (GO: 0009987), biological regulation (GO: 0065007), metabolic process (GO: 0008152), and response to stimulus (GO: 0050896). Interestingly, the terms of reproduction (GO: 0000003) and reproductive process (GO: 0022414) were also enriched. Several genes enriched in the reproduction GO term are involved in the pantothenate biosynthesis pathway (Supplementary Figure S2), which has previously been identified as possibly relevant to foetal development [Citation18], and was predicted to be relevant for daughter fertility by the panther database. As for the protein class analysis, the most enriched GO terms were metabolite interconversion enzyme (PC00262), transmembrane signal receptor (PC00197), and gene-specific transcriptional regulator (PC00264). Additionally, binding (GO: 0005488) and catalytic activity (GO: 0003824) were the two most enriched GO terms under molecular function. The functional classification of these genes is shown in the supplementary file 5.
Figure 3. GO term enrichment analysis of the genes that contain at least one DMR (q < 0.05). The biological process (yellow), the molecular function (green), and protein class (grey) GO term summary from GO slim analysis in panther. GO: Gene Ontology. By default, only the term that presents a p-value lower than 0.05 is displayed.
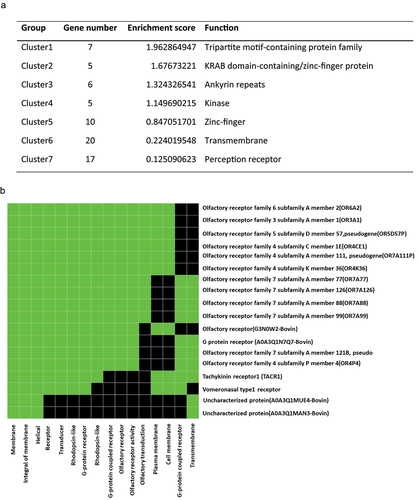
In order to analyse in detail how DNA methylation profiles may relate to gene expression, we next explored the functions of the 791 genes that covered at least one significant DMC (q < 0.05) (Supplementary file 4). Based on the biological functions of the genes, seven distinct functional clusters were identified (), and the corresponding genes were listed in Supplementary file 4. Based on the enrichment score, the tripartite motif-containing (TRIM) protein family had the highest enrichment. There has been evidence that the TRIM gene family plays an essential role in spermatogenesis and embryogenesis. For example, Trim27 was shown to regulate meiosis by regulating the formation of the XY body and the proliferation of germ cells during spermatogenesis [19]; and Trim28 played a vital role in the demethylation of DNA in embryos, preventing developmental abnormalities [Citation19]. Our analysis also identified a significant group enrichment of the olfactory receptor genes (). In recent studies, olfactory factors in sperm have been hypothesized to function as chemosensors to detect chemicals in the female genital tract [Citation20], several olfactory receptors in human sperm are involved in Ca2+ signalling, which affects capacitation [Citation21].
Figure 4. The functional cluster analysis. a. The functional cluster enrichment of the genes contained at least one significant CG (q < 0.05), and the enrichment scores were calculated based on the ranks of the biological significance of gene groups according to overall with the high stringency EASE scores of all enriched annotation terms. The gene list was first run with DAVID functional annotation chart to get the p-value (EASE score) for each enriched annotation term and then calculated the geometric mean of EASE scores of those terms involved in this gene group. b. The significantly enriched group of the olfactory receptors contained at least one significant CG. The green colour represents corresponding gene-term associations that were positively reported. The black colour represents corresponding gene-term associations not reported yet.
Signaling pathway analysis and network prediction
Comparatively to rodents and humans, bovine’s signalling pathways have been less investigated, and many uncharacterized genes have been identified in our study (genes that covered DMRs), therefore, we performed network prediction analysis using Ingenuity pathway analysis (IPA) and genes containing at least one DMC (q < 0.05). Upon analysis, the top associated network was found to be related to respiratory disease, cell morphology, and embryonic development (). As shown in , the embryonic development process was also considerably enriched in the physiology system development and function analysis.
Table 1. Ingenuity pathway analysis (IPA) analysis of genes that covered at least one differentially expressed cytosine (DMC) (q < 0.05) between high daughter fertility and low daughter fertility groups.
Table 2. A list of the top canonical physiological function was generated via IPA.
The correlation between the bovine sperm and daughter fertility
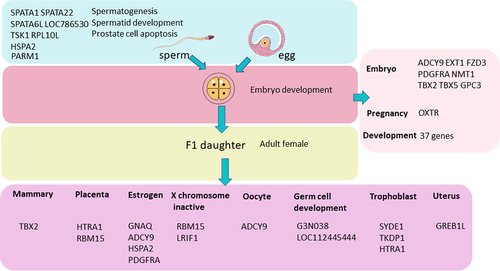
In order to produce a fertile daughter, the zygote must be able to develop after fertilization and the recipient cow must become pregnant successfully. In light of this, a collection of genes associated with sperm, embryo development, and the female reproductive system were identified based on genes’ GO annotations, and genes covered at least one DMR were adopted (q < 0.05, ). shows eight genes related to sperm fertility and 46 genes related to pregnancy, development (Supplementary File 5), and embryos. Sixteen genes are linked to adult female reproduction. Moreover, comparing our results with our recently published paper on bull fertility-associated methylation markers [Citation15] (Supplementary Figure D3), bull fertility and daughter fertility shared 15.9% of genes that covered at least one DMR (q < 0.05), which could be classified into seven clusters according to their function (Supplementary file 3).
Figure 5. Genes associated with daughter fertility. All the genes covered at least one significant CG (q < 0.05), and the genes that were identified and shown in the figure are based on the gene annotation. By searching the keywords sperm, embryo, reproduction, development, female, placenta, and oestrogen in the annotation of all the genes that covered at least one DMR, the corresponding genes listed in Figure 5 were identified.
Discussion
Identifying biomarkers in genetics and epigenetics to determine superior bulls in early life before artificial insemination was required due to the decline in dairy cattle fertility. The paternal sperm-derived DMC and DMR associated with daughter fertility phenotype are described in this study. In our recently published review paper [Citation11], we summarized that the methylation ‘escapee’ or ‘re-establish’ model might be involved in this correlation, and worth further exploring. The enzyme-based whole-genome methylation sequencing was applied to semen from high and lower daughter fertility groups of the bulls in this study [Citation22]. In contrast to RRBS (Reduced Representation Bisulfite Sequencing), which targets the CCGG regions, does not inform about other genome regions [Citation23], the platform we adopted in this study allowed us to include the methylation status of all the CpG sites in the analysis, including all the low CpG-density regions such as intergenic ‘gene deserts’ regions. Further, the 25× sequence coverage generated significant statistical power, and enzyme conversion enabled lower bias than bisulphite conversion. It is also noteworthy that random additive effect and random male impact had been included in the algorithms of the daughter fertility index calculation [Citation24,Citation25] which avoid confounding factor impact.
DMCs and DMRs were discovered mostly on the X chromosome during the DNA methylation marker discovery phase of this study (). This could be explained by the fact that X-type sperm can be passed from father to daughter, and therefore, more methylation information on the X chromosome is associated with daughter fertility. Our study found 27 genes that contained at least one DMR on the X chromosome, including six unknown genes (Supplementary file 6). Three clusters were identified based on the biological functions of these genes. The most enriched cluster was associated with germ cell development (enrichment score = 2.139) (Supplementary file 6). Besides, the X chromosome genes identified in this study included lipoma HMGIC fusion partner-like 1 (LHFPL1), Y-linked-like protein-like (LOC526488), glypicans 3 (GPC3), ARL14 effector protein-like pseudogene (LOC112441485), histone H2B ½ (LOC523458), serine-rich and transmembrane domain containing 2 (SERTM2), small integral membrane protein 10 (SMIM10), interleukin 1 receptor accessory protein-like 1 (IL1RAPL1), melanoma-associated antigen 10-like (LOC618806) and Zinc finger protein 75D (ZNF75D). Taking some genes as examples, there is high expression of ARL14ep in the ovary, testis, and uterus [Citation26], and one SNP at ARL14ep was strongly associated with rs11031005, which was correlated with the Follicle stimulating hormone (FSH) concentration [Citation27]. Testis-specific histone H2B ½ is associated with spermatogenesis, gamete formation, and chromosome alignment. Its expression is higher in bull sperm with poor fertility [Citation27] . There is some evidence that SERTM2 is involved in establishing and developing the corpus luteum of sheep [Citation28]. SMIM10 was significantly regulated by luteinizing hormone in thecal cells and was downregulated after the Luteinising hormone (LH) surge [Citation29]. Interleukin 1 receptor accessory protein is expressed in the human endometrium throughout the menstrual cycle [Citation30], and ZNF75D was differentially methylated in mammary tissue after in-utero heat stress [Citation31,Citation32].
The functional classification of the genes (covered DMCs) evidences some interesting information. For example, the most enriched cluster is the Tripartite motif-containing protein family () which was considered essential for embryo and spermatogenesis, for example, Trim28 prevents the embryo from development abnormality [Citation19] and Trim27 [Citation31]could regulate meiosis during spermatogenesis.
Interestingly, the highly enriched sensory perception receptors (), including olfactory and tachykinin receptors, indicated a link to daughter fertility as well which could be reflected in their essential role in chemotaxis and capacitation [Citation20]. Tachykinins are expressed in the female reproductive tract, and tachykinins and tachykinin receptors are also present in the male testis [Citation31], their possible mechanisms in regulating fertility involve the control of Gonadotropin-releasing hormone (GnRH) secretion, the regulation of sperm motility or the inhibition of testosterone production [Citation33].
Based on the annotation of the genes (Supplementary file 5) that contained at least one DMR (q < 0.05), we found that many reproduction and development-associated genes could be identified and grouped, as shown in . Eight genes, namely, Spermatogenesis Associated 1 (SPATA1), Spermatogenesis Associated 22 (SPATA22), Spermatogenesis Associated 6 Like (SPATA6L), spermatogenesis associated 1 pseudogene (LOC786530), Testis associated actin remodelling kinase 1 (TESK1), ribosomal protein L10 like (RPL10L), heat shock protein family A (Hsp70) member 2 (HSPA2) and prostate androgen-regulated mucin-like protein 1 (PARM1) were associated with spermatid development, spermatogenesis or the prostate cell apoptosis process based on their annotation. Interestingly, 37 genes were correlated with the development process (Supplementary file 5). Eight genes, including Adenylate cyclase 9 (ADCY9), Platelet-derived growth factor receptor alpha (PDGFRA), and N-myristoyl transferase 1 (NMT1) were associated with in-utero embryonic development, and Exostosin glycosyltransferase 1 (EXT1), frizzled class receptor 3 (FZD3), T-box transcription factor 2 (TBX2), T-box transcription factor 5 (TBX5), and glypican 3 (GPC3) were related to embryo development (Supplementary file 5). Oxytocin receptor (OXTR) is linked to ovarian and uterine functions. Besides, according to the gene annotation exported from DAVID tools, eight gene groups linked with the female reproductive system were mined (). The details of these genes can be found in supplemental file 5. All the genes’ functions were determined solely by their annotation, with no predictions based on literature mining. As a result, the methylation status of the genes listed in was linked to daughter fertility. Using a RRBS platform, Miriama et al [Citation7] recently found that sperm-associated DMCs and DMRs are associated with bull fertility, covered by seven unique genes (SFRP1, STXBP4, BCR, PSMG4, ARSG, ATP11A, RXRA), which are involved in spermatogenesis and early embryonic development. Comparing our result with their result, we found that Sortilin Related VPS10 Domain Containing Receptor 3 (SORCS3) overlapped.
Thanks to the insemination data accumulated in the Canadian dairy network (CDN), we obtained high-quality experimental material with daughter fertility record phenotypes. Furthermore, compared to humans and other experimental animals, artificial insemination procedures were employed in dairy herds, which excluded the intercourse impact that usually matters in other models as well. In the future, the identified differential DNA methylation patterns could be combined with the genetic tools that have been developed in recent years to provide functional tools to improve dairy cattle fertility and the profitability of dairy farms. The integrated analysis of the results in this study and the bull fertility biomarker identification study [Citation15] could also add new knowledge on epigenetic intergenerational inheritance in domestic animals. The pathways and differentially methylated genes we identified also provide a good reference for future dairy cattle fertility-related studies.
In this study, there were several points worth noting. First, the phenotype-daughter fertility has a low heritability might explain the individual differences, secondly, one straw of semen may not represent all semen samples of the bull [Citation34], we are possibly missing other sites that may vary more from ejaculate to ejaculate. Thirdly, a larger number of bulls and the specific genes must be tested to validate the sensitivity and specificity of the selected methylation markers. Our results will complement the existing predicting tool and enhance the effectiveness of bull selection, as mentioned in the introduction, while the effect of management and nutrition on fertility cannot be underestimated, especially when the heritability is low.
In summary, the signature of differential DNA methylated regions and differentially methylated cytosines were identified to be associated with daughter fertility in bovine, which will have a great potential to be used as a complementary breeding tool for superior bull (have better daughter fertility performance) determination in the future, our result does suggest that the paternal sperm exist epigenetic information that associated with female offspring phenotype. Besides, the X chromosome of sperm appears to be responsible for the differences in DNA methylation associated with daughter fertility. Furthermore, the enriched sensory receptors found in this study indicate that paternal sperm contributes to fertility in female offspring.
Materials and methods
Chemical reagents
All reagents in this study were of tissue culture grade and obtained from Sigma-Aldrich (Oakville, Ontario, Canada).
Bull data and sample selection
Semex Alliance (Guelph, ON, Canada), a livestock genetics company, provided a pre-filtered 100-bull list that includes Holstein and Jersey, each bull produced at least 6000 doses of semen up to 30 January 2019. The daughter fertility index (DFI), which is presently utilized by the Canadian dairy industry to improve female fertility [Citation35], was used for sire determination. The DFI is a combination trait that considers different reproduction traits together and is based on the vast artificial insemination (AI) database accumulated by the Canadian Dairy Network (CDN), six traits were included in the daughter fertility index, namely, age at first service (0.11), the 56-day non-return rate for heifers (0.16) (cows) (0.34), the interval from calving to the first service (0.15), the interval from insemination to conception (heifer (0.08) and cow (0.16)). The mean of the DFI in the provided bull list is 103.41 and does not follow the normal distribution since all the semen in this study had been used for commercial purposes (only high daughter fertility and less daughter fertility sires were included), thus the extreme value of the DFI in each contrast was determined. The six bulls with the highest (mean = 112.6) and six bulls with the lowest (mean = 92) daughter fertility were identified and selected for comparison. Frozen semen from these two groups of bulls was used for genomic DNA extraction as described below.
Sperm genomic DNA extraction
Half of the semen straw (125 uL) for each bull was thawed in water at 37°C for 1 min. Sperm cells were then washed with 1.0 mL phosphate-buffered saline (PH7.4). After centrifugation at 1550 g for 2 min to pellet the sperm cell, the supernatant was aspirated and 1 mL of somatic cell lysis buffer (0.5% Triton X-100 in phosphate-buffered saline) was added to the pellet and incubated for 10 min on ice. The further extraction protocol was downloaded previously (https://www.qiagen.com/jp/resources/download.aspx?id=c26b2186-076f-48cc-ad01-a4e07b26ab1d&lang=en), the DNeasy Blood & Tissue kit from QIAGEN (Toronto, ON, Canada, Cat. #69504) was used and the manipulation was following the user developed protocol. Briefly, after centrifugation at 1550 g for 5 min, the pellets were resuspended in 100 µL phosphate buffered saline in a microcentrifuge tube and 100 µL buffer X2 (Buffer X2:20 mM Tris. Cl (pH 8.0), 20 mM EDTA (PH 8.0), 200 mM NaCl, 4% SDS) was added, followed by 80 mM dithiothreitol and 12.5 µL/ml Proteinase K. The mixture was then incubated in a thermomixer at 56°C overnight until the sample was dissolved. 200 uL buffer AL was added to the samples and mixed thoroughly by vortexing. 200 uL ethanol (96%-100%) was added and mixed thoroughly by vortexing. After pipetting the mixture into the DNeasy Mini spin column and centrifuging at 8000 rpm for 1 min, the flow-through was discarded. The spin column was then put in a new collection tube. 500uL buffer AW1 was added and the tube was centrifuged for 1 min at 8000 rpm. The flow through was discarded, 500 uL buffer AW2 was added and the tube was centrifuged for 3 min at 14,000 rpm to dry the DNeasy membrane. Then, discard the flow through and the collection tube. The column was put in a new microcentrifuge tube, and 50 uL buffer AE was directly loaded onto the DNeasy membrane. Incubate the column at room temperature for 1 min and then centrifuge for 1 min at 8000 rpm to elute the DNA. Subsequently, the concentration (Supplementary file 1, sheet 2) and DNA integrity were measured with the 2200 Tape Station (Agilent Technologies) system, and all centrifugation steps were carried out at room temperature (15–25°C).
Library preparation and enzyme-based sequencing
The extracted genome DNA was sent to Genome Quebec for library construction and sequencing. Briefly, NEBNext® Enzymatic Methyl-seq Kit (E7120L) and EM-seq workflow [Citation36] was adopted to perform the whole genome methylation sequencing. This method results in less DNA fragmentation and damage than traditional bisulphite sequencing [Citation36]. After the end repair of the fragmented extracted DNA, the methylated adapter was ligated to the DNA. The enzymatic conversion was then performed following the mechanism that the Apolipoprotein B mRNA-editing (APOBEC) enzyme converted the unmethylated cytosine into uracil, while Tet Methylcytosine Dioxygenase 2 (TET2) and an oxidation enhancer protected the methylated cytosine. During PCR amplification, the uracil was replaced by thymine and methylated cytosine was unchanged. Finally, the sequencing was performed on the Illumina HiSeq X sequencing platform to yield the 150 bp paired end reads with a 25X coverage.
Data analysis
Adapter removal and quality trimming were performed using TRIMMOMATIC (0.36). We used TRIMMOMATIC [Citation37] in Paired-End Mode. Illumina adapters were identified using seed matches allowing for two mismatches. Matches are then extended and clipped to remove the adapter if a score of 30 is reached. N bases and trailing below a quality score of 30 were removed as well as the first 4 bases of each reads. Finally, reads smaller than 25 bases long after these steps were filtered out. BISMARK [Citation38] (0.22.1) was then used to align reads from the libraries to a synthetic converted version of the reference bovine genome (ARS-UCD1.2, https://www.ncbi.nlm.nih.gov/assembly/GCF_002263795.1/). Bismark was run with Bowtie 2 [Citation38,Citation39] against the bisulphite genome and we set the maximum insert size to 1000bp. The enzyme conversion rate was calculated by the imprinting genes Potassium Voltage-Gated Channel Subfamily Q Member 1 (KCNQ1), Small Nuclear Ribonucleoprotein Polypeptide N (SNRPN), and H19 via using Bismark. The Methyl-Kit R [Citation37] package was used to identify differentially methylated bins and differentially methylated cytosines.CG sites that showed a methylation difference higher than 20% (q < 0.01) are considered DMC, and bins that present a methylation difference of no less than 15% (q < 1.1e-26) are deemed promising DMRs. The high-daughter fertility group was set as the treatment group and the low-daughter fertility group was set as the control for comparison. The reads with lower than 10 X coverage were removed for CG analysis. In addition, considering that the single CG has fewer phenotype effects than regions, sliding window analyses were performed using a window size of 500 bp and a step of 100 bp to identify differentially methylated regions, and the regions that contained at least 10 CG were selected. No overdispersion correction was done and logistic regression was adopted for statistical analysis given that multiple replicates per group. P-values were adjusted to q-values using the SLIM method [Citation40]. The annotation of the DMC and DMR was done by using the annotate peak function of the ChIPseeker R package [Citation41] via overlapping the methylkit output CSV file with the GFF3 files that were downloaded from the ensemble (http://ftp.ensembl.org/pub/release-104/gff3/bos_taurus/Bos_taurus.ARS-UCD1.2.104.gff3.gz). The genes that contain at least one DMC or DMR are defined as differentially methylated genes.
Gene ontology enrichment, pathway analysis and functional classification analysis
Gene ontology slim enrichment was performed by PANTHER [Citation42] with the genes that contain at least one DMR (q < 0.05). Ingenuity pathway analysis (IPA) is mainly based on uncategorized human, rat, and mouse information, networks representing molecular interaction were constructed based on the IPA [Citation43] database. Since less pathways and mechanisms have been explored in bovines compared to rodents and humans, the gene cluster analysis based on biological functions was done in the Database for Annotation, Visualization, and Integrated Discovery (DAVID) [Citation44]. The genes that covered at least one DMCs (q < 0.05) were uploaded into DAVID for functional classification analysis, the functional classification tool generates a gene-to-gene similarity matrix based on shared functional annotation using thousands of annotation terms from 12 functional annotation categories, and the low classification stringency was adopted in this study.
Abbreviations
| AI | = | (Artificial Insemination) |
| CDN | = | (Canadian dairy network) |
| DMCs | = | (Differentially methylated cytosines) |
| DMRs | = | (Differentially methylated regions) |
| DMGs | = | (Differentially methylated genes) |
| DFI | = | (Daughter fertility index) |
| DNA | = | (Deoxyribonucleic acid) |
| DAVID (Database for Annotation | = | (Visualization, and Integrated Discovery) |
| IPA | = | (Ingenuity pathway analysis) |
| WGBS | = | (Whole genome bisulphite sequencing) |
| NRR | = | (Non-return rate) |
| GO | = | (Gene ontology) |
| KEGG | = | (Kyoto Encyclopedia of Genes and Genomes) |
| NRR | = | (Non-Return rate) |
| QTLs | = | (Quantitative trait locus) |
| SNP | = | (Single-nucleotide Polymorphism) |
| SPATA1 | = | (Spermatogenesis Associated 1) |
| SPATA6L | = | (Spermatogenesis Associated 6 Like) |
| SPATA22 | = | (Spermatogenesis Associated 22) |
| APOBECA | = | (Apolipoprotein B mRNA-editing) |
| SncRNA | = | (Small non-coding RNA) |
| LOC786530 | = | (Spermatogenesis associated 1 pseudogene) |
| TESK1 | = | (Testis associated actin remodelling kinase 1) |
| RPL10L | = | (Ribosomal protein L10 like) |
| HSPA2 | = | (Heat shock protein family A (Hsp70) member 2) |
| PARM1 | = | (Prostate androgen-regulated mucin-like protein 1) |
| UTR | = | (Untranslated regions) |
| ADCY9 | = | (Adenylate cyclase 9) |
| PDGFRA | = | (Platelet derived growth factor receptor alpha) |
| NMT1 | = | (N-myristoyltransferase 1) |
| EXT1 | = | (Exostosin glycosyltransferase 1) |
| FZD3 | = | (Frizzled class receptor 3) |
| TBX2 | = | (T-box transcription factor 2) |
| IPA | = | (Ingenuity pathway analysis) |
| KCNQ1 | = | (Potassium Voltage-Gated Channel Subfamily Q Member 1) |
| SNRPN | = | (Small Nuclear Ribonucleoprotein Polypeptide N) |
| TBX5 | = | (T-box transcription factor 5) |
| GPC3 | = | (Glypican 3) |
| OXTR | = | (Oxytocin receptor) |
Availability of data and materials
Whole-genome methylation sequencing data were deposited in NCBI’s Gene Expression Omnibus (GEO) and are accessible through the GEO series accession number: GSE211926.
Supplemental Material
Download Zip (508.7 KB)Acknowledgments
This work was supported by SEMEX Alliance (Guelph, ON, Canada) and the CHINA SCHOLARSHIP COUNCIL. The authors thank SEMEX for the donation of the experimental materials and the use of their private database. The authors would also like to thank Patrick Blondin and collaborators from Boviteq (Saint-Hyacinthe, QC, Canada) for the original data collection, Julien Prunier and Eric Fournier for the bioinformatic analysis, Dominic Gagné for technical support, and Sylvie Bilodeau-Goeseels for proofreading the manuscript.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/15592294.2023.2280889
Additional information
Funding
References
- Berry DP, Friggens NC, Lucy M, et al. Milk production and fertility in cattle, Annu. Rev Anim Biosci. 2016;4:269–13. doi: 10.1146/annurev-animal-021815-111406
- Thundathil JC, Dance AL, Kastelic JP. Fertility management of bulls to improve beef cattle productivity. Theriogenology. 2016;86:397–405. doi: 10.1016/j.theriogenology.2016.04.054
- Liu A, Wang Y, Sahana G, et al. Genome-wide association studies for female fertility traits in Chinese and Nordic holsteins, Sci. Sci Rep. 2017;7:8487. doi: 10.1038/s41598-017-09170-9
- Cai Z, Guldbrandtsen B, Lund MS, et al. Prioritizing candidate genes for fertility in dairy cows using gene-based analysis, functional annotation and differential gene expression. BMC Genomics. 2019;20: doi: 10.1186/s12864-019-5638-9
- Peter S, Ekstrm TJ. Epigenetics–a helpful tool to better understand processes in clinical nephrology?, Nephrol. Dial. Transplant Off Publ Eur Dial Transpl Assoc - Eur Ren Assoc. 2008;23:1493. doi: 10.1093/ndt/gfn056
- Kitamura A, Miyauchi N, Hamada H, et al. Epigenetic alterations in sperm associated with male infertility, Congenit. Anom. 2015;55:133–144. doi: 10.1111/cga.12113
- Štiavnická M, Chaulot-Talmon A, Perrier J-P, et al. Sperm DNA methylation patterns at discrete CpGs and genes involved in embryonic development are related to bull fertility. BMC Genomics. 2022;23:379. doi: 10.1186/s12864-022-08614-5
- Kropp J, Carrillo JA, Namous H, et al. Male fertility status is associated with DNA methylation signatures in sperm and transcriptomic profiles of bovine preimplantation embryos. BMC Genomics. 2017;18:280. doi: 10.1186/s12864-017-3673-y
- Atsem S, Reichenbach J, Potabattula R, et al. Paternal age effects on sperm FOXK1 and KCNA7 methylation and transmission into the next generation. Hum Mol Genet. 2016;25:4996–5005. doi: 10.1093/hmg/ddw328
- Jenkins TG, Aston KI, Pflueger C, et al. Age-associated sperm DNA methylation alterations: possible implications in offspring disease susceptibility. PLoS Genet. 2014;10:e1004458. doi: 10.1371/journal.pgen.1004458
- Zhang Y, Sirard M-A. Epigenetic inheritance of acquired traits through DNA methylation. Anim Front Rev Mag Anim Agric. 2021;11:19–27. doi: 10.1093/af/vfab052
- Painter RC, Osmond C, Gluckman P, et al. Transgenerational effects of prenatal exposure to the Dutch famine on neonatal adiposity and health in later life, BJOG int. J Obstet Gynaecol. 2008;115:1243–1249. doi: 10.1111/j.1471-0528.2008.01822.x
- Burdge GC, Hoile SP, Uller T, et al. Progressive, transgenerational changes in offspring phenotype and Epigenotype following nutritional transition. PLoS One. 2011;6:e28282. doi: 10.1371/journal.pone.0028282
- Zhang Y, Chaput C, Fournier E, et al. Comparing the whole genome methylation landscape of dairy calf blood cells revealed intergenerational inheritance of the maternal metabolism. Epigenetics. 2022;17:705–714. doi: 10.1080/15592294.2021.1955188
- Zhang Y, Bruna de Lima C, Labrecque R, et al. Whole genome DNA methylation analysis of the sperm in relation to bull fertility, Reprod. Camb Engl. 2023;165:REP-22–0283. doi: 10.1530/REP-22-0283
- Doormaal BJV, Kistemaker G, Fatehi J, et al. Genetic Evaluation of Female Fertility in Canadian Dairy Breeds[J]. 2004;32.
- Guo YS, Tao JZ. Metabolomics and pathway analyses to characterize metabolic alterations in pregnant dairy cows on D 17 and D 45 after AI. Sci Rep. 2018;8:5973. doi: 10.1038/s41598-018-23983-2
- Zhuang X-J, Tang W-H, Feng X, et al. Trim27 interacts with Slx2, is associated with meiotic processes during spermatogenesis. Cell Cycle Georget Tex. 2016;15:2576–2584. doi: 10.1080/15384101.2016.1174796
- Messerschmidt DM, Vries WD, Ito M, et al. Trim28 is required for epigenetic stability during mouse oocyte to embryo transition. Science. 2012;335:1499–1502. doi: 10.1126/science.1216154
- Spehr M, Schwane K, Riffell JA, et al. Odorant receptors and olfactory-like signaling mechanisms in mammalian sperm, Mol. Cell Endocrinol. 2006;250:128–136. doi: 10.1016/j.mce.2005.12.035
- Flegel C, Vogel F, Hofreuter A, et al. Characterization of the olfactory receptors expressed in human spermatozoa. Front Mol Biosci. 2015 73;2. 10.3389/fmolb.2015.00073
- Feng S, Zhong Z, Wang M, et al. Efficient and accurate determination of genome-wide DNA methylation patterns in Arabidopsis thaliana with enzymatic methyl sequencing. Epigenetics Chromatin. 2020;13:42. doi: 10.1186/s13072-020-00361-9
- Meissner A, Gnirke A, Bell GW, et al. Reduced representation bisulfite sequencing for comparative high-resolution DNA methylation analysis. Nucleic Acids Res. 2005;33:5868–5877. doi: 10.1093/nar/gki901
- Doormaal B, Kistemaker G, Fatehi J, et al. Genetic Evaluation of female fertility in Canadian dairy breeds. 2004.
- Jamrozik J, Fatehi J, Kistemaker GJ, et al. Estimates of Genetic Parameters for Canadian Holstein Female Reproduction Traits. J Dairy Sci. 2005;88:2199–2208. doi: 10.3168/jds.S0022-0302(05)72895-2
- Kutchy NA, Velho A, Menezes ESB, et al. Testis specific histone 2B is associated with sperm chromatin dynamics and bull fertility-a pilot study, Reprod. Biol Endocrinol RBE. 2017;15:59. doi: 10.1186/s12958-017-0274-1
- Ruth KS, Campbell PJ, Chew S, et al. Genome-wide association study with 1000 genomes imputation identifies signals for nine sex hormone-related phenotypes. Eur J Hum Genet EJHG. 2016;24:284–290. doi: 10.1038/ejhg.2015.102
- Liu A, Liu M, Li Y, et al. Differential expression and prediction of function of lncRnas in the ovaries of low and high fecundity hanper sheep, Reprod. Domest Anim Zuchthyg. 2021;56:604–620. doi: 10.1111/rda.13898
- Christenson LK, Gunewardena S, Hong X, et al. Research resource: preovulatory LH surge effects on follicular theca and granulosa transcriptomes, Mol. Endocrinol Baltim Md. 2013;27:1153–1171. doi: 10.1210/me.2013-1093
- Bigonnesse F, Marois M, Maheux R, et al. Interleukin-1 receptor accessory protein is constitutively expressed in human endometrium throughout the menstrual cycle, Mol. Hum Reprod. 2001;7:333–339. doi: 10.1093/molehr/7.4.333
- Skibiel AL, Peñagaricano F, Amorín R, et al. In Utero Heat Stress Alters the Offspring Epigenome, Sci. Sci Rep. 2018;8:14609. doi: 10.1038/s41598-018-32975-1
- Pinto FM, Almeida TA, Hernandez M, et al. Candenas, mRNA expression of tachykinins and tachykinin receptors in different human tissues. Eur J Pharmacol. 2004;494:233–239. doi: 10.1016/j.ejphar.2004.05.016
- Blasco V, Pinto FM, González-Ravina C, et al. Tachykinins and kisspeptins in the regulation of human male fertility. J Clin Med. 2019;9:113. doi: 10.3390/jcm9010113
- Lambert S, Blondin P, Vigneault C, et al. Spermatozoa DNA methylation patterns differ due to peripubertal age in bulls. Theriogenology. 2018;106:21–29. doi: 10.1016/j.theriogenology.2017.10.006
- Walsh SW, Williams EJ, Evans ACO. A review of the causes of poor fertility in high milk producing dairy cows. Anim Reprod Sci. 2011;123:127–138. doi: 10.1016/j.anireprosci.2010.12.001
- Vaisvila R, Ponnaluri VKC, Sun Z, et al. Davis, enzymatic methyl sequencing detects DNA methylation at single-base resolution from picograms of DNA. Genome Res. 2021;31:1280–1289. doi: 10.1101/gr.266551.120
- Bolger AM, Marc L, Bjoern U. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–2120. doi: 10.1093/bioinformatics/btu170
- Krueger F, Andrews SR. Bismark: a flexible aligner and methylation caller for bisulfite-seq applications, Bioinforma. Oxf Engl. 2011;27:1571–1572. doi: 10.1093/bioinformatics/btr167
- Langmead B, Salzberg SL. Fast gapped-read alignment with bowtie 2. Nat Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923
- Wang H-Q, Tuominen LK, Tsai C-J. SLIM: a sliding linear model for estimating the proportion of true null hypotheses in datasets with dependence structures, Bioinforma. Oxf Engl. 2011;27:225–231. doi: 10.1093/bioinformatics/btq650
- Yu G, Wang L-G, He Q-Y. ChIPseeker: an R/Bioconductor package for ChIP peak annotation, comparison and visualization, Bioinforma. Oxf Engl. 2015;31:2382–2383. doi: 10.1093/bioinformatics/btv145
- Mi H, Thomas P. PANTHER pathway: an ontology-based pathway database coupled with data analysis tools, methods Mol. Biol Clifton NJ. 2009;563:123–140. doi: 10.1007/978-1-60761-175-2_7
- Krämer A, Green J, Pollard J, et al. Causal analysis approaches in Ingenuity pathway analysis, Bioinforma. Oxf Engl. 2014;30:523–530. doi: 10.1093/bioinformatics/btt703
- Huang DW, Sherman BT, Tan Q, et al. The DAVID gene functional classification tool: a novel biological module-centric algorithm to functionally analyze large gene lists. Genome Biol. 2007;8:R183. doi: 10.1186/gb-2007-8-9-r183