
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
Foeniculum vulgare Mill. commonly known as fennel, is a globally recognized aromatic medicinal plant and culinary herb with widespread popularity due to its antimicrobial, antioxidant, carminative, and diuretic properties, among others. Although the phenotypic effects of salinity stress have been previously explored in fennel, the molecular mechanisms underlying responses to elevated salinity in this plant remain elusive. MicroRNAs (miRNAs) are tiny, endogenous, and extensively conserved non-coding RNAs (ncRNAs) typically ranging from 20 to 24 nucleotides (nt) in length that play a major role in a myriad of biological functions. In fact, a number of miRNAs have been extensively associated with responses to abiotic stress in plants. Consequently, employing computational methodologies and rigorous filtering criteria, 40 putative miRNAs belonging to 25 different families were characterized from fennel in this study. Subsequently, employing the psRNATarget tool, a total of 67 different candidate target transcripts for the characterized fennel miRNAs were predicted. Additionally, the expression patterns of six selected fennel miRNAs (i.e. fvu-miR156a, fvu-miR162a-3p, fvu-miR166a-3p, fvu-miR167a-5p, fvu-miR171a-3p, and fvu-miR408-3p) were analyzed under salinity stress conditions via qPCR. This article holds notable significance as it identifies not only 40 putative miRNAs in fennel, a non-model plant, but also pioneers the analysis of their expression under salinity stress conditions.
1. Introduction
MicroRNAs (miRNAs) are small, endogenous, highly conserved non-coding RNAs (ncRNAs) of approximately 20 to 24 nucleotides (nt) in length.Citation1 They exert a pivotal control on the post-transcriptional regulation of gene expression by virtue of their capability to mediate the degradation or translational inhibition of messenger RNAs (mRNAs).Citation1 As a matter of fact, plant miRNAs have been widely associated with several biological functions, including development and reproduction,Citation2 cell reprogramming,Citation3 secondary metabolism,Citation4 and biotic and abiotic stress responses.Citation5
The initiation of miRNA biogenesis in plants begins at the nucleus with the transcription of miRNA genes (MIRs) by the enzyme RNA polymerase II (RNAPII), giving rise to long primary transcripts known as primary-miRNAs (pri-miRNAs). Successive processing involves the cleavage of pri-miRNAs into stem-loop RNA precursors (pre-miRNAs) through the action of DICER-LIKE1 (DCL1), HYPONASTIC LEAVES 1 (HYL1), and SERRATE (SE) enzymes.Citation6,Citation7 Following this, DCL1 recognizes and cleaves the hairpin loop of the pre-miRNAs, generating miRNA/miRNA* duplexes (where miRNA refers to the guide strand and miRNA* to the passenger strand). Eventually, the guide strand of the mature miRNA duplex associates with the RNA-Induced Silencing Complex (RISC), forming a miRNA-ribonucleoprotein complex guided by the ARGONAUTE (AGO) protein. This complex effectively interacts with mRNA targets, culminating in the regulatory processes associated with miRNA-mediated gene silencing.Citation6,Citation7
The pervasive adoption of small RNA high-throughput sequencing (sRNA-seq) technology has accelerated a notable surge in the identification of miRNAs over the past decade. Concurrently, there has been an apparent escalation in the entries within the miRBase registry, underscoring the expanding repository of documented plant miRNAs.Citation8 Besides, the evolutionary conservation of numerous plant miRNAs facilitates the streamlined characterization of miRNA orthologs in novel plant species through the documentation of homologous sequences.Citation9 Even so, reliance solely on sequence-based in silico homology approaches for the identification of putative miRNAs in new plants may generate false-positive outcomes. Therefore, a comprehensive approach should be adopted, considering not only the primary sequence but also the secondary structures of pre-miRNAs, along with critical parameters such as length, GC content, Minimum Folding Free Energy (MFE), and Minimum Folding Free Energy Index (MFEI). This multifaceted consideration is essential to enhance the precision of computational predictions, enabling the discrimination of miRNAs from other coding RNA or ncRNA entities.Citation10 Nevertheless, it is strongly advisable to validate the computationally predicted miRNAs through experimental methods.Citation11
Fennel (Foeniculum vulgare Mill.), an enduring tropical herb belonging to the Apiaceae (also known as Umbelliferae) family and native to the Mediterranean region, is distinguished by its abundant branches, tender leaves, small yellow flowers, elongated fruits, fragrant scent, and a foliage resembling fine hairs.Citation12,Citation13 Moreover, different parts of fennel plant (e.g., seeds, fruits, and leaves) own remarkable antispasmodic, carminative, diuretic, antimicrobial, and antioxidant properties, among others,Citation14 due to the presence of several bioactive compounds such as limonene, chlorogenic acid, glycosides of quercetin, rosmarinic acid, estragole, anethole, and kaempferol.Citation15
Salinity stress represents a major abiotic factor exerting considerable impact on the growth, development, and productivity of plants, as well as on crop yields. In this context, it has been demonstrated that salinity stress affects biomass, photosynthetic pigments, leaf area, and relative water content in F. vulgare.Citation16,Citation17 Despite the above, the biological significance of miRNAs in the context of salinity stress in F. vulgare remains enigmatic. Therefore, understanding the regulatory mechanisms through which miRNAs operate under salinity stress conditions in this species holds significance for unraveling the molecular intricacies of stress response pathways. Consequently, in this current investigation, the published draft genome sequence of fennel (GenBank assembly accession GCA_003724115.2) facilitated the characterization of numerous miRNAs and their respective targets. Additionally, an analysis of the expression patterns of some of these miRNAs under salinity stress was conducted, aiming to enhance our comprehension of the physiological role played by miRNAs in fennel.
2. Materials and methods
2.1. Computational prediction of potential fennel miRNAs and their putative pre-miRNAs
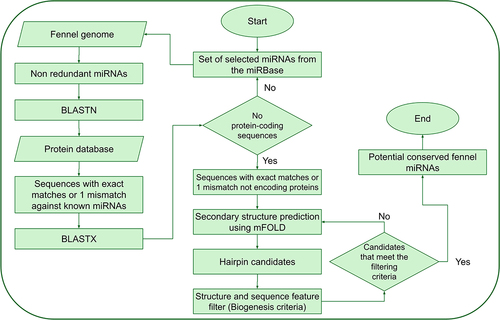
An in silico analysis was conducted to predict and identify potential miRNAs in fennel using a reference set of plant miRNAs sourced from the miRBase database.Citation18 The reference set comprised a total of 1,216 known mature miRNAs from Arabidopsis thaliana (428), Glycine max (756), and Panax ginseng (32). In brief, the known miRNAs were subjected to BLASTn against the fennel genome, and sequences with exact matches or a maximum of 1 mismatch were manually selected. Potential precursor sequences of approximately 400 nt in length (200 nt upstream and 200 nt downstream of the BLAST hit region) were identified, with the exclusion of protein-coding sequences. The selected precursors’ stable secondary structures were generated using the mFold web server,Citation19 and their stability was assessed through established stringent filtering criteria. These criteria included ensuring that (a) the pre-miRNA possessed a stem-loop structure containing the mature miRNA sequence within one arm, (b) the mature miRNA was not presented in the terminal loop of the hairpin structure, (c) the mature miRNA had fewer than nine mismatches with the opposite miRNA* sequence, (d) and the potential stem-loop candidate exhibited a minimum negative MFE (or ΔG, −kcal/mol) coupled with a high MFEI (the MFEI calculation followed the formula provided below). The procedure for the computational prediction of miRNAs in fennel is illustrated in .
Figure 1. Schematic depiction of the fennel miRNA prediction procedure.
2.2. Phylogenetic and conservation analysis of fennel miRNA precursors
For the conservation analysis of the predicted fennel pre-miRNA candidates, the miRNA precursor sequences from various plant species were recovered,Citation18 including Amborella trichopoda (atr), Arabidopsis lyrata (aly), Arabidopsis thaliana (ath), Asparagus officinalis (aff), Brachypodium distachyon (bdi), Brassica napus (bna), Carica papaya (cpa), Citrus sinensis (csi), Cucumis melo (cme), Fragaria vesca (fve), Glycine max (gma), Linum usitatissimum (lus), Malus domestica (mdm), Nicotiana tabacum (nta), Oryza sativa (osa), Theobroma cacao (tcc), and Vitis vinifera (vvi). After that, multiple sequence alignment and phylogenetic tree assembly (grounded in the Tamura-Nei model with 100 boot-strapped replicates) were executed using the Molecular Evolutionary Genetics Analysis (MEGA) software (version 11.0.11).Citation20 Besides, the sequence conservation analysis of three representative predicted fennel pre-miRNAs, i.e., pre-miR156a, pre-miR166a, and pre-miR171a, was carried out using the WebLogo tool,Citation21 considering their orthologs. Furthermore, to clarify the highly conserved characteristics of miRNAs across diverse species and their potential for cross-species transferability, a syntenic map was created employing the putative fennel pre-miRNAs against the well-annotated Daucus carota (carrot) genome (GenBank assembly accession GCF_001625215.1), a closely related species of fennel.Citation22,Citation23
2.3. Target prediction of fennel miRNAs and their functional annotations
In order to predict the potential target transcripts of the fennel miRNAs, the “Plant Small RNA Target Analysis Server” (psRNATarget)Citation24 was utilized. The search for target transcripts was conducted against the database of D. carota (that belongs to the same family of fennel, i.e., ApiaceaeCitation25,Citation26) due to the unavailability of the fennel database on the psRNATarget cDNA library. The specified selection parameters included an expectation value of 3, translation inhibition ranges spanning from 9 to 11 nt, a top target count of 10, a penalty for G:U pair of 0.5, and allowance for 1.5 mismatches in the seed region. Afterward, the protein data for the identified sequences was retrieved using UniProt BLAST. Subsequently, the gene ontology (GO) analysis of the putative fennel targets was performed, elucidating the associated biological processes, cellular components, and molecular functions for each GO term employing the QuickGO tool.Citation27 Additionally, to discern the co-regulation among potential targets, a biological network was constructed using Minimum Free Energy (MFE) values of miRNA-target interactions and visualized through the Cytoscape 3.10.1 software.Citation28 Lastly, KEGG analysis was executed via the KEGG Automatic Annotation Server (KAAS)Citation29 to explore the metabolic pathways and networks modulated by the prospective fennel miRNAs, utilizing the Bi-directional Best Hit (BBH) method.
2.4. Plant materials, stress treatment, small RNA extraction, and miRNA expression analysis
Fennel seeds were germinated in sterile paper towel at 25°C in darkness and the seedlings were then transferred into two hydroponic systems with automated LED illumination from iDOO (ID-IG301, Eastvale, CA, USA) containing a culture media prepared by mixing distilled water with a registered commercial hydroponic solution (Interagro, Guadalajara, JAL, Mexico) with a pH adjusted to 7.Citation30 The plants were allowed to grow under controlled environmental conditions at 25°C and with a 16 h photoperiod (automatically regulated by the hydroponic system) for 6 weeks; the hydroponic solutions were renewed weekly. Following this, the hydroponic solution for one of the systems was formulated with a concentration of 200 mM NaCl to induce salinity stress, whereas the solution for the other system was prepared without NaCl, serving as the control.
Next, leaves from both stressed and control fennel plants were harvested after a short exposure (24 h) and a long exposure (72 h). Small RNA molecules (<200 nt) were extracted from the leaf tissues using the miRNeasy Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer’s guidelines. Subsequently, both the quality and quantity of the RNA samples were assessed using Nanodrop One (Thermo Scientific, Wilmington, NC, USA). Thereafter, 1 µg of RNA from the individual samples was polyadenylated (using a modified oligo dT primer) and reverse transcribed, employing the mRQ Buffer and enzyme of the Mir-X miRNA First-Strand Synthesis kit (Takara, Tokyo, Japan). The qRT-PCR experiment was conducted utilizing the Step One Real-Time PCR System (Applied Biosystems, Carlsbad, CA, USA) and the Mir-X miRNA TB Green qRT-PCR kit (Takara, Tokyo, Japan).
The entire predicted miRNA sequence of each of the analyzed miRNAs served as the forward primer, while the adapter-specific mRQ3’ primer provided with the kit was used as the reverse primer. Each reaction was prepared in a 12.5 µL volume, comprising 1× SYBR Advantage Premix, 1× ROX dye, 0.2 µM of both forward and reverse primers, and 2 µL of the first-strand cDNA. A total of six selected miRNAs (i.e., fvu-miR156a, fvu-miR162a-3p, fvu-miR166a-3p, fvu-miR167a-5p, fvu-miR171a-3p, and fvu-miR408-3p), previously documented to play pivotal roles in both biotic and abiotic stress responsesCitation31 were chosen for the qRT-PCR experiment. The qPCR conditions comprised an initial denaturation at 95°C for 10 s, 45 cycles of denaturation at 95°C for 5 s, annealing at 63°C for 20 s, and concluding with a dissociation curve consisting of 95°C for 30 s, 55°C for 20 s, and 95°C for 20 s. The relative fold change values were calculated through the application of the comparative Ct method, also known as the delta-delta Ct method (2−ΔΔCT). All the qPCR reactions were performed using three biological replicates and two technical replicates.
3. Results
3.1. Characterization of fennel miRNAs and their candidate precursors
In this investigation, employing computational strategies and sticking to rigorous filtering criteria, a set of 40 potential miRNAs distributed across 25 families was identified (). The predominant length among the identified fennel miRNAs was 21 nt. The precursors of these fennel miRNAs exhibited substantial variability in size, ranging from 68 to 210 nt, with an average length of 114 ± 38 nt. The fennel miRNA fvu-miR319c displayed the longest precursor with 210 nt, whereas fvu-miR167a-5p exhibited the shortest one with 68 nt. Besides, the MFE values of the precursors ranged from −88.40 to −30.00 kcal/mol, with an average of −47.27 ± 13.87 kcal/mol. Concurrently, the MFEI values varied from 0.71 to 1.47, averaging 1.03 ± 0.19. The predicted secondary structures of fennel miRNA precursors featuring higher MFEI values (top 10) are depicted in .
Figure 2. Secondary stem-loop structures of the top ten putative fennel pre-miRNAs. The corresponding mature miRNAs are indicated in red font.
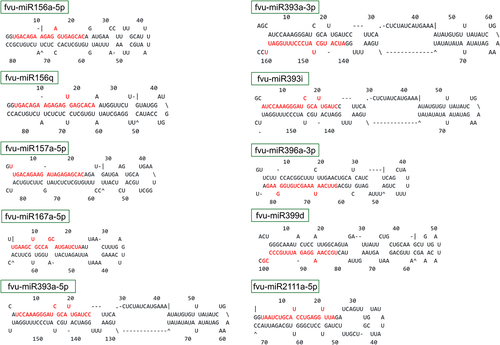
Table 1. Candidate miRNAs identified in fennel through bioinformatic methods.
3.2. Conservation analysis of fennel miRNAs and their putative precursors
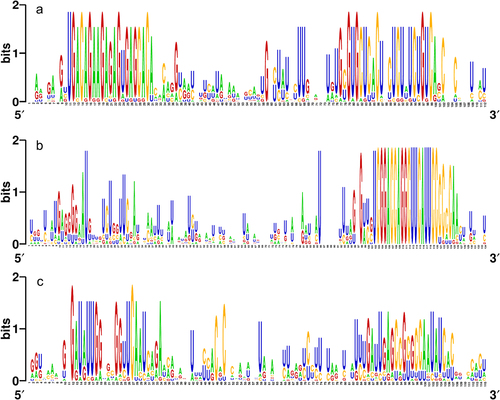
The evolutionary relationships among miRNAs can be explored due to the conserved nature of pre-miRNAs and mature miRNAs. As a matter of fact, search approaches for homologous sequences rely on known miRNAs and pre-miRNAs as references.Citation32 As depicted in , high conservation of sequence was observed among the orthologs of a set of representative fennel pre-miRNAs comprised of pre-miR156a, pre-miR166a, and pre-miR171a.
Figure 3. Graphical representation (WebLogo) of the conserved nucleotide sequences among the pre-miRNA orthologs of (a) pre-miR156a, (b) pre-miR166a, and (c) pre-miR171a.
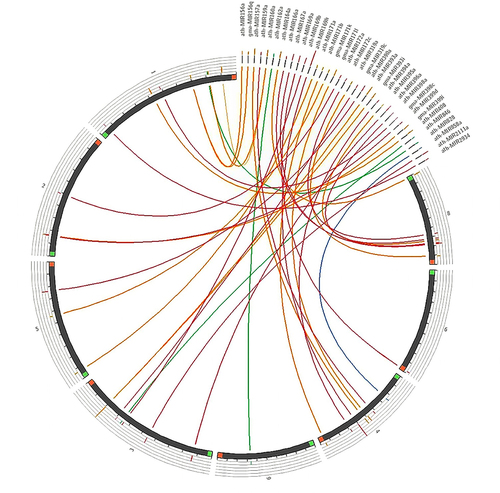
Additionally, the phylogenetic analysis of the set of representative pre-miRNAs suggested that fvu-miR156a is closely related to the miRNA orthologs present in melon (cme-miR156a), asparagus (aof-miR156a), tobacco (nta-miR156a), and rice (osa-miR156a); while fvu-pre-miR171a is closer to papaya, common grape vine, and amborella (cpa-miR171a, vvi-miR171a, and atr-miR171a). Additionally, fvu-miR166a showed a close phylogenetic relationship with Brachypodium distachyon (bdi-miR166a), cacao (tcc-miR166a), wild strawberry (fve-miR166a), amborella (atr-miR166a), and melon (cme-miR166a) (). On the other hand, the comparative synteny map demonstrated the broad distribution of the prospective fennel miRNA orthologs within the carrot genome, illustrating their ability to transfer across species during evolutionary processes ().
Figure 4. Phylogenetic assessment was conducted on the putative fennel miRNAs fvu-miR156a, fvu-miR166a, and fvu-miR171a (emphasized within the red squares) using their predicted precursor sequences.
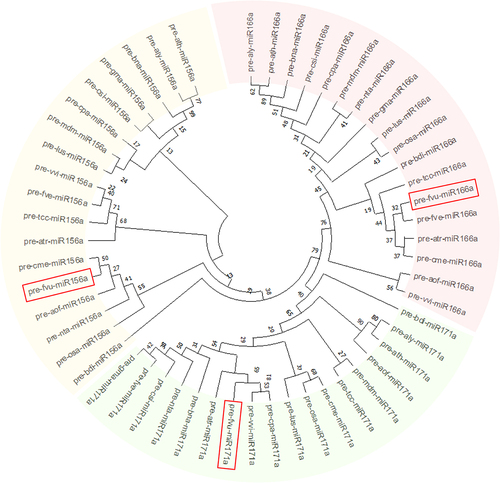
Figure 5. Synteny map comparing candidate fennel miRNAs with the well-annotated genome of a closely related species, carrot (D. carota).
3.3. Predicted targets for fennel miRNAs and their functional annotations
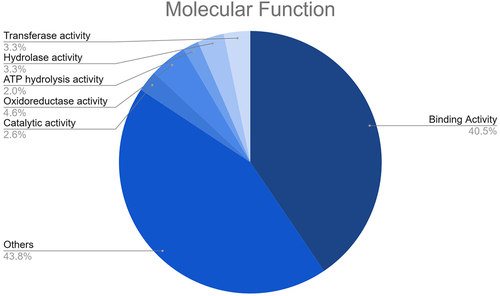
In this study, 67 different potential target transcripts of fennel miRNAs were predicted. Such target transcripts include a number of important molecular entities, such as SBP-type domain-containing proteins, HTH myb-type domain-containing proteins, transcription factor GAMYB-like, auxin response factors, NAC domain-containing proteins, F-box domain-containing proteins, magnesium transporter, alpha-glucosidase, cytochrome P450, ceramidase, among others. Subsequently, a Gene Ontology (GO) analysis of the predicted targets was performed to gain a more profound insight into the plausible functions of the fennel miRNAs. As well, this analysis helped to unravel the regulatory network of the miRNA genes involved in biological mechanisms, molecular functions, and cellular components (). The GO enrichment analysis indicated that the fennel miRNA targets are involved in diverse molecular functions, including binding activity (e.g., DNA binding, protein binding, and metal ion binding), transferase activity, hydrolase activity, ATP hydrolysis activity, oxidoreductase activity, and catalytic activity ().
Figure 6. Results of the GO enrichment analysis of the putative fennel miRNA targets related to Molecular Function.
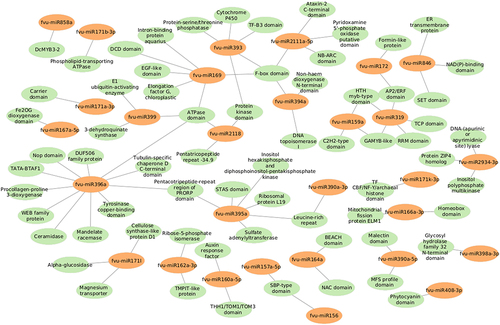
These targets were also found to participate in a variety of biological processes like regulation of DNA-templated transcription, RNA modification, response to other organism, defense response, and transmembrane transport (). Meanwhile, the most enriched cellular components were found to be the membrane, nucleus, cellulose synthase complex, endoplasmic reticulum membrane, endoplasmic reticulum, nucleolus, cytosol, cytoplasm, amongst others (). Besides, the KEGG analysis implied that the putative fennel miRNA targets are involved in 30 different metabolic pathways, being the “Phosphatidylinositol signaling system” the most enriched one (). Additionally, the gene network analysis illustrated in shows the coordinated regulation of multiple potential fennel miRNA target genes.
Figure 7. Results of the GO enrichment analysis of the putative fennel miRNA targets related to Biological Process.
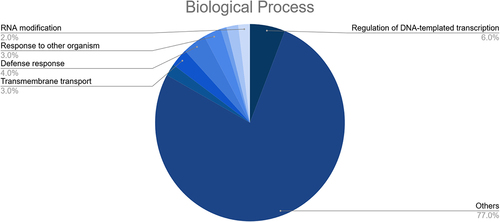
Figure 8. Results of the GO enrichment analysis of the putative fennel miRNA targets related to Cellular Component.
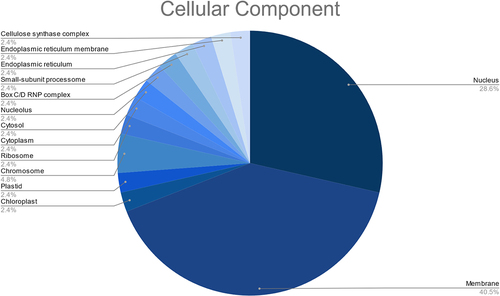
Figure 9. KEGG pathways of the predicted fennel miRNA targets that were mapped using the KAAS tool.
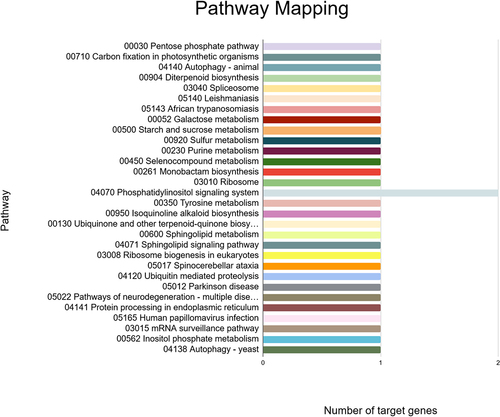
Figure 10. Network interactions based on minimum free energy (MFE) between prospective fennel miRNAs (orange circles) and their corresponding potential targets (green circles).
3.4. Expression profiling of fennel miRNAs under salinity stress
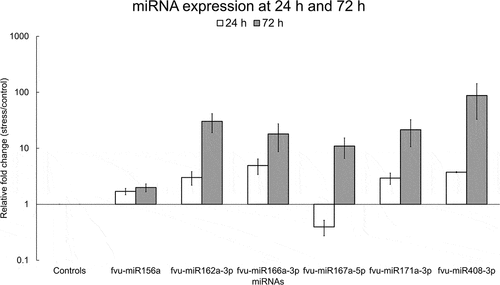
To explore the impact of salinity stress on the expression of selected fennel miRNAs (fvu-miR156a, fvu-miR162a-3p, fvu-miR166a-3p, fvu-miR167a-5p, fvu-miR171a-3p, and fvu-miR408-3p) in leaves, a qRT-PCR experiment was conducted. The results revealed differential expression of all six miRNAs in response to salinity stress both at 24 h and 72 h. The expression levels of fvu-miR156a, fvu-miR162a-3p, fvu-miR166a-3p, fvu-miR171a-3p, and fvu-miR408-3p were upregulated at 24 h, while fvu-miR167a-5p was downregulated when compared to the fennel plants that were not subjected to salinity stress. Later, at 72 h, the expression levels of all the studied miRNAs were found to be upregulated ().
Figure 11. Relative fold change of selected fennel miRNAs under salinity stress at 24 h and 72 h. The fold change was calculated using the delta-delta CT method, U6 snRNA was utilized as the normalization control, and the values were normalized relative to the control condition (set as 1). Error bars indicate the standard error of the biological replicates.
4. Discussion
The significant functions of miRNAs in plant development, growth, and adaptation to both biotic and abiotic stress have turned them into compelling molecular subjects of research, especially in the pursuit of miRNA-based strategies for plant improvement.Citation33 Despite the fact that miRNAs have been extensively investigated in various key crops and model plants, such as Arabidopsis Citation34 and soybean,Citation35 the functional roles of these master regulators of gene expression remain elusive in non-model plant species like fennel. However, recent advances in the assembly of the fennel genomeCitation26 and leaf transcriptomeCitation25 have allowed us to advance in the prediction and functional characterization of the miRNAs belonging to this aromatic medicinal plant. In fact, the putative fennel miRNAs identified in this current investigation, as well as their corresponding precursors, presented high sequence conservation with their orthologs from different monocot and dicot plant species. This finding suggests that miRNAs are universally conserved between monocotyledonous and dicotyledonous species,Citation36 implying that they may fulfill similar physiological functions.
The precursors of the fennel miRNAs predicted herein exhibited a wide range of sizes, spanning from 68 to 210 nt and displayed stable stem-loop secondary structures, consistent with findings reported in numerous other recently studied non-model plant species including cranberry,Citation37 neem,Citation38 and Sorghum bicolor.Citation39 Remarkably, all the predicted precursor structures of the fennel miRNAs showed high MFEI values that ranged from 0.71 to 1.47, with an average of 1.03, which is greater than the MFEI values associated with other RNA species (e.g., mRNAs: 0.62–0.66, tRNAs: 0.64, or rRNAs: 0.59).Citation40 In our assessment, a notable predominance of uracil at the initial position of the predicted miRNAs was also acknowledged. This observation reinforces the reliability of our results and aligns with previous findings emphasizing the pivotal role of uracil at the start of the mature miRNA in governing miRNA-mediated regulatory mechanisms.Citation41,Citation42
In line with previous findings, the majority of predicted targets of fennel miRNAs are transcription factors primarily associated with plant growth, developmental patterning, cell differentiation, and stress responses.Citation43 Several of the predicted transcripts targeted by the putative fennel miRNAs corresponded to transcription factors SBP/SPL, which have fundamental participation in flowering time, changes from the vegetative phase to the reproductive phase, and juvenile-to-adult transition. In concordance with our results, SBP/SPL transcription factors are reported to be targeted by the miR156 family, mainly by miR156 and miR157.Citation44 In addition, MYB transcription factors were predicted as targets of fennel miRNAs. These transcription factors play crucial roles in regulating various functions, including the regulation of gene expression in diverse pathways that include secondary metabolism, plant hormone signaling, as well as developmental and morphological processes.Citation45 Indeed, as herein noticed with the fennel miRNAs, members belonging to the miR159 family regulate developmental phase transitions in plants by targeting genes encoding MYB transcription factors.Citation46
Another important group of transcription factors that were detected to be targeted by fennel miR160a-5p are comprised of ARFs; these transcripts play a major role in both auxin biosynthesis and auxin signaling pathway, being key participants in processes like leaf, flower and fruit development, as well as diverse stress responses.Citation47 Meanwhile, NAC domain-containing proteins were identified as the plausible targets of fvu-miR164a, which largely agrees with the fact that the miR164-NAC module is widely conserved in plant species and has a crucial role in adaptative stress responses.Citation48 These findings shed light on the potential biological significance of fennel miRNAs, indicating the conservation of such functions across species and highlighting transcription factors as their primary targets.
Interestingly, plants possess specialized mechanisms to mitigate the effects of salinity stress, with miRNAs playing a pivotal role in their regulation. Mechanistically, salt stress can induce the expression (upregulation) of miRNAs, which in turn downregulate target mRNAs encoding proteins that have negative effects on the plant’s response to high salinity. Contrarywise, the downregulation of other miRNAs leads to the accumulation of their target mRNAs, which positively contribute to stress adaptation.Citation49 In this context, our results show that fvu-miR156a and fvu-miR162a-3p were upregulated under salinity conditions both at 24 h and 72 h, while fvu-miR167a-5p was upregulated at 72 h. These observations agree with the outcomes reported in previous articles. For instance, Sun et al.Citation50 elucidated that miR156, miR162, and miR167 (among other miRNAs) were upregulated in switchgrass under higher salt concentrations. As well, it has been demonstrated that the transgenic expression of zma-miR156 enhanced salt and osmotic tolerance in tobacco via downregulating NtSPL2 and NtSPL9.Citation51 thus indicating that the upregulation of miR156 is highly relevant in some plant species to overcome salinity stress. Likewise, Luo et al.Citation52 detected that miR162 was upregulated at 24 h and 48 h in Poa pratensis callus subjected to salt stress. Moreover, miR167 was detected to be upregulated under salinity stress conditions in Tamarix chinensis, causing the downregulation of ARFs such as TcARF6.Citation53
On the other hand, fvu-miR166a-3p was upregulated in fennel plants that were subjected to salinity stress. Although it has been widely reported that diverse members of the miR166 family are downregulated under salinity stress in several plants, including guavaCitation54 and Arabidopsis,Citation55 other reports on soybeanCitation56 and highbush blueberryCitation57 imply that some members of the same miRNA family become upregulated as a result of salt stress. Furthermore, as acknowledged with fvu-miR171a-5p, Liu et al.Citation58 and Deng et al.Citation59 stated that miR171 is upregulated during salinity stress in Arabidopsis and barley, respectively. In regard to miR408, Ma et al.Citation60 unveiled that this miRNA becomes upregulated under high salinity conditions in Arabidopsis and that its overexpression enhances the tolerance to salinity stress. Such observations coincide with the fact that fvu-miR408-3p was also upregulated in fennel after the induction of salinity stress. In fact, the overexpression of miR408 encloses a prospective use in plant improvement since transgenic tobacco plants expressing the Salvia miltiorrhiza miR408 displayed enhanced seed germination and decreased accumulation of reactive oxygen species under salt stress conditions.Citation61 Overall, the findings discussed in the previous lines shed light on the crucial role of fennel miRNAs in response to salinity stress. However, further exploration is required to define the complex miRNA-mRNA interactions and extensive regulatory networks that occur in this medicinal plant.
5. Conclusions
The regulatory roles of miRNAs at the post-transcriptional level significantly influence the overall gene regulatory network in plants, and the study of the interplay between miRNAs and their corresponding mRNA targets in non-model plants could significantly benefit the progress in the understanding of miRNAs’ biological functions. Over the last years, a growing interest in the computational identification of miRNAs has emerged due to the availability of advanced bioinformatic tools and sequence resources in public databases. This study represents one of the first analyses of miRNAs and their targets in fennel, where 40 potential miRNAs from 25 families were identified using rigorous filtering criteria and homology-based approaches. Additionally, 67 candidate target transcripts were identified for the predicted fennel miRNAs, providing insights into the regulatory roles of these master regulators of gene expression. Notably, six of the predicted miRNAs were confirmed through qPCR experiments, validating their existence and expression in fennel under both control and salinity stress conditions. As well, the current study on the effects of salinity stress on the expression levels of the selected fennel miRNAs enhances our understanding of their functional roles under stress conditions. It is worth noting that the application of artificial miRNA technology and CRISPR/Cas-based methods targeting miRNAs for crop improvement have shown promising results, and this communication also paves the way for future research on salinity stress tolerance in fennel by potentially using such technologies. Overall, this report contributes to the understanding of miRNA-mediated regulatory mechanisms in fennel and provides a foundation for future research on crop enhancement via applying miRNA-based strategies.
6. Future perspectives
It is worth emphasizing that fennel is a plant that has been relatively underexplored in terms of its genetic makeup,Citation26 with only a limited number of studies on its genetic composition and even fewer investigations focused on the non-coding regions of its genome. For instance, Attia et al.Citation62 elucidated that, under salinity stress conditions, spermine synthase 1 (SPMS1), ornithine decarboxylase 1 (ODC1), and arginine decarboxylase (ADC1) were upregulated in fennel hypocotyls and cotyledons, while S-adenosylmethionine decarboxylase 1 (SAMDC1) was downregulated in fennel radicle. Moreover, when fennel seedlings were subjected to a treatment combination of salt and gibberellic acid (GA), ODC1 was the only gene overexpressed in hypocotyls and cotyledons, whereas ADC1 was upregulated in hypocotyls, cotyledons, and radicle.Citation62 Although the aforementioned study did not delve into the molecular mechanisms underlying the observed changes in gene expression, the widely reported regulatory roles of miRNAs in salt stress responses and phytohormone signaling suggest the necessity of investigating the molecular association of miRNAs with stress responses against high salinity in fennel, as well as the interplay between fennel miRNAs, GA perception, and signal transduction pathway.
As previously mentioned, fennel represents a highly abundant source of health-promoting bioactive compounds. Consequently, comprehending the elements that regulate secondary metabolism pathways at the transcriptome level could be of utmost importance for effectively manipulating the genetic makeup of fennel and enhancing the production of phytochemicals endowed with therapeutic properties. In this regard, at least three fennel genes have been reported to participate in the biosynthesis of t-anethole, one of the most representative components of fennel essential oils with antimicrobial and anticancer activities.Citation63–65 These genes are associated with transcripts encoding the enzymes t-anol/isoeugenol synthase (IGS1), t-anol/isoeugenol O-methyltransferase (AIMT1), and phenylalanine ammonia-lyase (PAL).Citation25,Citation66 Particularly, Abdollahi et al.Citation66 reported that PAL and IGS1 exhibited significant upregulation 24 hours post-treatment with methyl jasmonate, coinciding with an elevation in the production of t-anethole. Consistently, these findings denote that conducting a thorough investigation into the miRNA-mediated regulation of transcripts encoding the IGS1, AIMT1, and PAL enzymes could help to develop miRNA-based methods to enhance the production of t-anethole in fennel. Remarkably, such an approach could also be extended to explore and improve other metabolic pathways existing within this plant.
Undoubtedly, the absence of a well-annotated genome for fennel poses challenges when predicting miRNA targets and, in this current research, the genome of carrot, a plant phylogenetically related to fennel, was used as a reference to predict miRNA targets in fennel. To overcome the limitation of a thoroughly annotated genome from fennel in forthcoming assays, various techniques can be employed to validate the presence of the putative fennel transcripts and proteins predicted herein. For instance, RNA-seq dataCitation67 specifically obtained from fennel tissues can confirm the expression of the forecasted miRNA targets. Similarly, techniques like qRT-PCRCitation68 can help to validate the expression levels of the predicted target genes. As well, mass spectrometry-based proteomics and other quantitative proteomic approachesCitation69 might be useful to detect and quantify the presence of target proteins in fennel tissues. In summary, these combined approaches could provide more reliable evidence regarding the interaction between miRNAs and their targets in fennel, even without a fully annotated genome.
Furthermore, it is crucial to acknowledge that a significantly underexplored aspect of fennel pertains to the transgenic approach for the investigation and enhancement of biological processes, encompassing miRNAs and other transcripts. Hence, the utilization of artificial miRNAs and the tailored engineering of MIRs through genome editing tools (e.g., CRISPR/Cas) hold great promise for deeper insights into the functions of fennel miRNAs and for generating plants with desired traits, including stress tolerance/resistance. In light of the information discussed throughout the previous sections, we strongly believe that the insights shared in this report will be relevant in laying the groundwork for the development of new research focused on analyzing both the coding and non-coding transcriptome of fennel.
Author contributions statement
Data analysis, writing, editing LABV; Experimental procedures, data collection and analysis LABV, MGO, SMF; Bioinformatics, and statistical analysis AS; Conceptualization, experimental design, critical writing, reviewing LABV and SP. All authors have read and agreed to the published version of the manuscript.
S1_Data of predicted fennel miRNA targets.xlsx
Download MS Excel (23.8 KB)Disclosure statement
No potential conflict of interest was reported by the author(s).
Supplementary Material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/15592324.2024.2361174
Additional information
Funding
References
- Begum Y. Regulatory role of microRNAs (miRNAs) in the recent development of abiotic stress tolerance of plants. Gene. 2022;821:146283. doi:10.1016/j.gene.2022.146283.
- Arnaud N, Laufs P. Plant miRNA integrated functions in development and reproduction. Front Plant Physiol. 2023;1:1271423. doi:10.3389/fphgy.2023.1271423.
- Bravo-Vázquez LA, Angulo‑Bejarano PI, Bandyopadhyay A, Sharma A, Paul S. Regulatory roles of noncoding RNAs in callus induction and plant cell dedifferentiation. Plant Cell Rep. 2023;42(4):689–14. doi:10.1007/s00299-023-02992-0.
- Owusu Adjei M, Zhou X, Mao M, Rafique F, Ma J. MicroRNAs roles in plants secondary metabolism. Plant Signal Behav. 2021;16(7):1915590. doi:10.1080/15592324.2021.1915590.
- Samynathan R, Venkidasamy B, Shanmugam A, Ramalingam S, Thiruvengadam M. Functional role of microRNA in the regulation of biotic and abiotic stress in agronomic plants. Front Genet. 2023;14:1272446. doi:10.3389/fgene.2023.1272446.
- Bajczyk M, Jarmolowski A, Jozwiak M, Pacak A, Pietrykowska H, Sierocka I, Swida-Barteczka A, Szewc L, Szweykowska-Kulinska Z. Recent insights into plant miRNA biogenesis: multiple layers of miRNA level regulation. Plants. 2023;12(2):342. doi:10.3390/plants12020342.
- Zhang L, Xiang Y, Chen S, Shi M, Jiang X, He Z, Gao S. Mechanisms of microRNA biogenesis and stability control in plants. Front Plant Sci. 2022;13:844149. doi:10.3389/fpls.2022.844149.
- Yu D, Wan Y, Ito H, Ma X, Xie T, Wang T, Shao C, Meng Y. PmiRDiscVali: An integrated pipeline for plant microRNA discovery and validation. BMC Genomics. 2019;20(1):133. doi:10.1186/s12864-019-5478-7.
- Hajieghrari B, Farrokhi N. Investigation on the conserved microRNA genes in higher plants. Plant Mol Biol Report. 2021;39(1):10–23. doi:10.1007/s11105-020-01228-9.
- Ayachit G, Shaikh I, Pandya H, Das J. Salient features, data and algorithms for microRNA screening from plants: a review on the gains and pitfalls of machine learning techniques. Curr Bioinform. 2020;15(10):1091–1103. doi:10.2174/1574893615999200601121756.
- Zhao Y, Kuang Z, Wang Y, Li L, Yang X. MicroRNA annotation in plants: current status and challenges. Brief Bioinform. 2021;22(5):bbab075. doi:10.1093/bib/bbab075.
- Jadid N, Widodo AF, Ermavitalini D, Sa’adah NN, Gunawan S, Nisa C. The medicinal Umbelliferae plant fennel (Foeniculum vulgare Mill.): Cultivation, traditional uses, phytopharmacological properties, and application in animal husbandry. Arabian J Chem. 2023;16(3):104541. doi:10.1016/j.arabjc.2023.104541.
- Rafieian F, Amani R, Rezaei A, Karaça AC, Jafari SM. Exploring fennel (Foeniculum vulgare): Composition, functional properties, potential health benefits, and safety. Crit Rev Food Sci Nutr. 2023;1–18. doi:10.1080/10408398.2023.2176817.
- Noreen S, Tufail T, Badar Ul Ain H, Awuchi CG. Pharmacological, nutraceutical, functional and therapeutic properties of fennel (Foeniculum vulgare). Int J Food Prop. 2023;26(1):915–927. doi:10.1080/10942912.2023.2192436.
- Maleš I, Pedisić S, Zorić Z, Elez-Garofulić I, Repajić M, You L, Vladimir-Knežević S, Butorac D, Dragović-Uzelac V. The medicinal and aromatic plants as ingredients in functional beverage production. J Funct Foods. 2022;96:105210. doi:10.1016/j.jff.2022.105210.
- Beyk-Khormizi A, Hosseini Sarghein S, Sarafraz-Ardakani MR, Moshtaghioun SM, Mousavi-Kouhi SM, Ganjeali A. Ameliorating effect of vermicompost on Foeniculum vulgare under saline condition. J Plant Nutr. 2023;46(8):1601–1615. doi:10.1080/01904167.2022.2092513.
- Mohammadi M, Pouryousef M, Farhang N. Study on germination and seedling growth of various ecotypes of fennel (Foeniculum vulgare Mill.) under salinity stress. J Appl Res Med Aromat Plants. 2023;34:100481. doi:10.1016/j.jarmap.2023.100481.
- Kozomara A, Birgaoanu M, Griffiths-Jones S. miRBase: From microRNA sequences to function. Nucleic Acids Res. 2019;47(D1):D155–D162. doi:10.1093/nar/gky1141.
- Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003;31(13):3406–3415. doi:10.1093/nar/gkg595.
- Tamura K, Stecher G, Kumar S. MEGA11: Molecular Evolutionary Genetics Analysis Version 11. Mol Biol Evol. 2021;38(7):3022–3027. doi:10.1093/molbev/msab120.
- Crooks GE, Hon G, Chandonia JM, Brenner SE. WebLogo: A sequence logo generator. Genome Res. 2004;14(6):1188–1190. doi:10.1101/gr.849004.
- Cholin SS, Poleshi CA, Manikanta DS, Christopher C. Exploring the genomic resources of carrot for cross-genera transferability and phylogenetic assessment among orphan spices and vegetables of Apiaceae family. Hortic Environ Biotechnol. 2019;60(1):81–93. doi:10.1007/s13580-018-0101-4.
- Youssef D, El-Bakatoushi R, Elframawy A, El-Sadek L, El Badan G. Molecular phylogenetic study of flavonoids in medicinal plants: a case study family Apiaceae. J Plant Res. 2023;136(3):305–322. doi:10.1007/s10265-023-01442-y.
- Dai X, Zhuang Z, Zhao PX. psRNATarget: a plant small RNA target analysis server (2017 release). Nucleic Acids Res. 2018;46(W1):W49–W54. doi:10.1093/nar/gky316.
- Palumbo F, Vannozzi A, Vitulo N, Lucchin M, Barcaccia G. The leaf transcriptome of fennel (Foeniculum vulgare Mill.) enables characterization of the t-anethole pathway and the discovery of microsatellites and single-nucleotide variants. Sci Rep. 2018;8(1):10459. doi:10.1038/s41598-018-28775-2.
- Palumbo F, Galla G, Vitulo N, Barcaccia G. First draft genome sequencing of fennel (Foeniculum vulgare Mill.): identification of simple sequence repeats and their application in marker-assisted breeding. Mol Breed. 2018;38(10):122. doi:10.1007/s11032-018-0884-0.
- Binns D, Dimmer E, Huntley R, Barrell D, O’Donovan C, Apweiler R. QuickGO: A web-based tool for gene ontology searching. Bioinformatics. 2009;25(22):3045–3046. doi:10.1093/bioinformatics/btp536.
- Cline MS, Smoot M, Cerami E, Kuchinsky A, Landys N, Workman C, Christmas R, Avila-Campilo I, Creech M, Gross B. et al. Integration of biological networks and gene expression data using Cytoscape. Nat Protoc. 2007;2(10):2366–2382. doi:10.1038/nprot.2007.324.
- Moriya Y, Itoh M, Okuda S, Yoshizawa AC, Kanehisa M. KAAS: an automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 2007;35(suppl_2):W182–W185. doi:10.1093/nar/gkm321.
- Fatima H, Hamdani SDA, Ahmed M, Rajput TA, Gul A, Amir R, Munir F, Malik SZ, Babar MM. Anti-MRSA potential of biogenic silver nanoparticles synthesized from hydroponically grown Foeniculum vulgare. Phytomed Plus. 2023;3(1):100415. doi:10.1016/j.phyplu.2023.100415.
- Pagano L, Rossi R, Paesano L, Marmiroli N, Marmiroli M. miRNA regulation and stress adaptation in plants. Environ Exp Bot. 2021;184:104369. doi:10.1016/j.envexpbot.2020.104369.
- Yu T, Xu N, Haque N, Gao C, Huang W, Huang Z. Popular computational tools used for miRNA prediction and their future development prospects. Interdiscip Sci Comput Life Sci. 2020;12(4):395–413. doi:10.1007/s12539-020-00387-3.
- Raza A, Charagh S, Karikari B, Sharif R, Yadav V, Mubarik MS, Habib M, Zhuang Y, Zhang C, Chen H. et al. miRNAs for crop improvement. Plant Physiol Biochem. 2023;201:107857. doi:10.1016/j.plaphy.2023.107857.
- Singh A, Gandhi N, Mishra V, Yadav S, Rai V, Sarkar AK. Role of abiotic stress responsive miRNAs in Arabidopsis root development. J Plant Biochem Biotechnol. 2020;29(4):733–742. doi:10.1007/s13562-020-00626-0.
- Chand Jha U, Nayyar H, Mantri N, Siddique KHM. Non-coding RNAs in legumes: their emerging roles in regulating biotic/abiotic stress responses and plant growth and development. Cells. 2021;10(7):1674. doi:10.3390/cells10071674.
- Choudhary A, Kumar A, Kaur H, Kaur N. MiRNA: the taskmaster of plant world. Biologia (Bratisl). 2021;76(5):1551–1567. doi:10.1007/s11756-021-00720-1.
- Chowdhury Paul S, Sharma A, Mehta R, Paul S. In silico characterization of microRNAs and their target transcripts from cranberry (Vaccinium macrocarpon). Cytol Genet. 2020;54(1):82–90. doi:10.3103/S0095452720010120.
- Paul S, Reyes-Pérez P, Angulo-Bejarano PI, Srivastava A, Ramalingam S, Sharma A. Characterization of microRNAs from neem (Azadirachta indica) and their tissue-specific expression study in leaves and stem. 3 Biotech. 2021;11(6):277. doi:10.1007/s13205-021-02839-z.
- Baqi A, Samiullah Rehman W, Bibi I, Menaa F, Khan Y, Albalawi DA, Sattar A. Identification and validation of functional miRNAs and their main targets in Sorghum bicolor. Mol Biotechnol. Published online December 28, 2023;1–15. doi:10.1007/s12033-023-00988-5.
- Khan QH. Identification of Conserved and Novel MicroRNAs with their Targets in Garden Pea (Pisum sativum L.) Leaves by High-Throughput Sequencing. Bioinform Biol Insights. 2023;17. doi:10.1177/11779322231162777.
- Xing H, Li Y, Ren Y, Zhao Y, Wu X, Li HL. Genome-wide investigation of microRNAs and expression profiles during rhizome development in ginger (Zingiber officinale Roscoe). BMC Genomics. 2022;23(1):49. doi:10.1186/s12864-021-08273-y.
- Bhavsar M, Mangukia N, Patel S, Rawal R, Mankad A. Unraveling the miRnome of Nicotiana rustica (Aztec tobacco) - A Genomewide computational assessment. Plant Gene. 2022;32:100378. doi:10.1016/j.plgene.2022.100378.
- Zhang F, Yang J, Zhang N, Wu J, Si H. Roles of microRNAs in abiotic stress response and characteristics regulation of plant. Front Plant Sci. 2022;13:919243. doi:10.3389/fpls.2022.919243.
- Dong Q, Hu B, Zhang C. microRNAs and their roles in plant development. Front Plant Sci. 2022;13:824240. doi:10.3389/fpls.2022.824240.
- Thakur S, Vasudev PG. MYB transcription factors and their role in medicinal plants. Mol Biol Rep. 2022;49(11):10995–11008. doi:10.1007/s11033-022-07825-z.
- Islam W, Tauqeer A, Waheed A, Zeng F. MicroRNA mediated plant responses to nutrient stress. Int J Mol Sci. 2022;23(5):2562. doi:10.3390/ijms23052562.
- Jodder J. miRNA-mediated regulation of auxin signaling pathway during plant development and stress responses. J Biosci. 2020;45(1):91. doi:10.1007/s12038-020-00062-1.
- Hernandez Y, Goswami K, Sanan-Mishra N. Stress induced dynamic adjustment of conserved miR164:NAC module. Plant-Environment Interact. 2020;1(2):134–151. doi:10.1002/pei3.10027.
- Lotfi A, Pervaiz T, Jiu S, Faghihi F, Jahanbakhshian Z, Khorzoghi EG, Fang J, Seyedi SM. Role of microRNAs and their target genes in salinity response in plants. Plant Growth Regul. 2017;82(3):377–390. doi:10.1007/s10725-017-0277-0.
- Sun G, Stewart CN, Xiao P, Zhang B. MicroRNA expression analysis in the cellulosic biofuel crop switchgrass (Panicum virgatum) under abiotic stress. PLoS One. 2012;7(3):e32017. doi:10.1371/journal.pone.0032017.
- Kang T, Yu CY, Liu Y, Song WM, Bao Y, Guo XT, Li B, Zhang HX. Subtly manipulated expression of Zmmir156 in tobacco improves drought and salt tolerance without changing the architecture of transgenic plants. Front Plant Sci. 2020;10:1664. doi:10.3389/fpls.2019.01664.
- Luo H, Zhou Z, Song G, Yao H, Han L. Antioxidant enzyme activity and microRNA are associated with growth of Poa pratensis callus under salt stress. Plant Biotechnol Rep. 2020;14(4):429–438. doi:10.1007/s11816-020-00620-x.
- Ye Y, Wang J, Wang W, Xu L. ARF family identification in Tamarix chinensis reveals the salt responsive expression of TcARF6 targeted by miR167. PeerJ. 2020;8(3):e8829. doi:10.7717/peerj.8829.
- Sharma A, Ruiz-Manriquez LM, Serrano-Cano FI, Reyes-Pérez PR, Tovar Alfaro CK, Barrón Andrade YE, Hernández Aros AK, Srivastava A, Paul S. Identification of microRNAs and their expression in leaf tissues of guava (Psidium guajava L.) under salinity stress. Agronomy. 2020;10(12):1920. doi:10.3390/agronomy10121920.
- Scintu D, Scacchi E, Cazzaniga F, Vinciarelli F, De Vivo M, Shtin M, Svolacchia N, Bertolotti G, Unterholzener SJ, Del Bianco M. et al. microRNA165 and 166 modulate response of the Arabidopsis root apical meristem to salt stress. Commun Biol. 2023;6(1):834. doi:10.1038/s42003-023-05201-6.
- Li H, Dong Y, Yin H, Wang N, Yang J, Liu X, Wang Y, Wu J, Li X. Characterization of the stress associated microRNAs in Glycine max by deep sequencing. BMC Plant Biol. 2011;11(1):170. doi:10.1186/1471-2229-11-170.
- Li Y, Wang X, Guo Q, Zhang X, Zhou L, Zhang Y, Zhang C. Conservation and diversity of miR166 family members from highbush blueberry (Vaccinium corymbosum) and their potential functions in abiotic stress. Front Genet. 2022;13:919856. doi:10.3389/fgene.2022.919856.
- Liu HH, Tian X, Li YJ, Wu CA, Zheng CC. Microarray-based analysis of stress-regulated microRNAs in Arabidopsis thaliana. RNA. 2008;14(5):836–843. doi:10.1261/rna.895308.
- Deng P, Wang L, Cui L, Feng K, Liu F, Du X, Tong W, Nie X, Ji W, Weining S. et al. Global identification of microRNAs and their targets in barley under salinity stress. PLoS One. 2015;10(9):e0137990. doi:10.1371/journal.pone.0137990.
- Ma C, Burd S, Lers A. miR408 is involved in abiotic stress responses in arabidopsis. Plant Journal. 2015;84(1):169–187. doi:10.1111/tpj.12999.
- Guo X, Niu J, Cao X. Heterologous expression of Salvia miltiorrhiza microRNA408 enhances tolerance to salt stress in Nicotiana benthamiana. Int J Mol Sci. 2018;19(12):3985. doi:10.3390/ijms19123985.
- Attia H, Alamer K, Algethami B, Zorrig W, Hessini K, Gupta K, Gupta B. Gibberellic acid interacts with salt stress on germination, growth and polyamine gene expression in fennel (Foeniculum vulgare Mill.) seedlings. Physiol Mol Biol Plants. 2022;28(3):607–622. doi:10.1007/s12298-022-01140-4.
- Gross M, Friedman J, Dudai N, Larkov O, Cohen Y, Bar E, Ravid U, Putievsky E, Lewinsohn E. Biosynthesis of estragole and t-anethole in bitter fennel (Foeniculum vulgare Mill. var. vulgare) chemotypes. Changes in SAM:phenylpropene O-methyltransferase activities during development. Plant Sci. 2002;163(5):1047–1053. doi:10.1016/S0168-9452(02)00279-0.
- Ghasemian A, Al-Marzoqi AH, Mostafavi SKS, Alghanimi YK, Teimouri M. Chemical composition and antimicrobial and cytotoxic activities of Foeniculum vulgare Mill essential oils. J Gastrointest Cancer. 2020;51(1):260–266. doi:10.1007/s12029-019-00241-w.
- Qiu J, Li H, Su H, Dong J, Luo M, Wang J, Leng B, Deng Y, Liu J, Deng X. Chemical composition of fennel essential oil and its impact on Staphylococcus aureus exotoxin production. World J Microbiol Biotechnol. 2012;28(4):1399–1405. doi:10.1007/s11274-011-0939-4.
- Abdollahi MR, Kianersi F, Moosavi SS, Dastan D, Asadi S. Identification and expression analysis of two genes involved in the biosynthesis of t-anethole in fennel (Foeniculum vulgare Mill.) and their up-regulation in leaves in response to methyl jasmonate treatments. J Plant Growth Regul. 2023;42(2):759–770. doi:10.1007/s00344-022-10583-8.
- Unamba CIN, Nag A, Sharma RK. Next generation sequencing technologies: the doorway to the unexplored genomics of non-model plants. Front Plant Sci. 2015;6:1074. doi:10.3389/fpls.2015.01074.
- Kern F, Backes C, Hirsch P, Fehlmann T, Hart M, Meese E, Keller A. What’s the target: Understanding two decades of in silico microRNA-target prediction. Brief Bioinform. 2020;21(6):1999–2010. doi:10.1093/bib/bbz111.
- Giambruno R, Mihailovich M, Bonaldi T. Mass spectrometry-based proteomics to unveil the non-coding RNA world. Front Mol Biosci. 2018;5:90. doi:10.3389/fmolb.2018.00090.