
Abstract
The development and introduction of injectable biomaterials and the identification of methods through which materials may form in situ are currently the topics of interest in materials science, specifically in the field of biomaterials. Over the last few decades, hydrogels which refers to the swellable polymeric matrices have gained wide attention due to their excellent characteristics such as swelling in different media, pH and temperature sensitivity, and sensitivity to other stimuli. Nowadays, injectable hydrogels have widely been studied due to their excellent insitu gelation at body temperature. These injectable insitu gels serve as depot system which ensures the local and systemic drug and gene delivery. These insitu gels also protect the proteins and peptide drugs invivo from environmental effect. The current review is made to report latest extensive literature regarding hydrogels, their classification, synthesis methods, structure of hydrogel network, methods of crosslinking, environment-sensitive hydrogel system, drug loading, and release, hydrogels as biosensors and applications of hydrogels.
1. Introduction
In recent years with current research in advanced drug delivery formulations, extensive efforts have been done to provide stable and more economical drug delivery systems. Off the various kinds of drug delivery systems which have been utilized as drug container, the main focus is on hydrogels drug delivery system, which are considered to not only reduce the problems associated with conventional dosage forms but also seems necessary for the novel drug delivery systems which need a convenient, stable, and biocompatible drug delivery system for molecules as small as NSAIDs (non-steroidal anti-inflammatory drugs) or as large as proteins and peptides.[Citation1,2]
Researchers have done extensive efforts to fabricate the physical and chemical properties of drug delivery systems to particularly regulate their permeability, biodegradability, surface functionality, biocompatibility, environmental response, and bio recognition sites to synthesize and obtain ‘intelligent’ drug delivery systems. Hydrogels have gained considerable interest and represent a drug delivery systems class that claimed as intelligent drug delivery.[Citation3,4]
Initial work on crosslinked HEMA hydrogels was performed in 1960 by Wichterle and Lim [Citation5] scientists have shown great interest in HEMA hydrogels due to their biocompatibility and hydrophilic nature.[Citation6–13]
Hydrogels refers to the drug delivery systems with crosslinked, three-dimensional hydrophilic networks that have the ability to swell but resist dissolving when placed in water or other biological fluid. Hydrogels can be synthesized in a various physical forms, including micro particles, nanoparticles, slabs, coatings, and films.[Citation14–16]
Hydrogels are water swollen polymeric matrices with three-dimensional configuration capable of absorbing high amounts of water or other biological fluids. Their tendency to swell, under physiological environment, makes them an intelligent and ideal material for biomedical applications.[Citation17]
The crosslinked network of the hydrogels contain large amount of hydrophilic moieties or groups. The presence of chemical residues such as carboxylic, sulfonic amidic, hydroxyl, primary amidic, and others that can be found within the polymer network backbone or as lateral chains mainly attributes to the hydrophilicity of the network. These chemical moieties form physical or chemical bonds between polymer network backbones. Water molecules penetrate the polymer networks, which leads to swelling and giving the hydrogels its original shape. In swollen state, hydrogels resembles soft and rubbery consistency like natural living tissues which is due to the presence of high water molecules in the network structure. The rubbery and soft character of the hydrogels has the advantage of reducing irritation to natural neighboring tissues.[Citation18] The three-dimensional structure of the hydrogels is due to the presence of crosslinks between the polymer chains. Due to the introduction of these crosslinks between the polymer chains, hydrogels tend to show pure elastic and sometime visco-elastic behavior.[Citation19] Crosslinking between the polymer chains can be provided by hydrogen bonding, vander Waals interactions, covalent bonds, or physical entanglements.[Citation20] The presence of crosslinks between the polymer chains renders these hydrophilic network structures to remain insoluble in an aqueous environment due to the hydrogen bonding and ionic interactions.[Citation18] For the preparation of hydrogels, numerous crosslinking methods have been used to introduce crosslink’s between the polymer chains. Since these hydrogels are biodegradable and biocompatible, they claim the presence of labile bonds in the network structure. These labile bonds in the polymer backbone have the tendency to break under physiological environment either through the action of enzymes, environmentally (such as temperature, pH, or electric field) or through chemical reactions mostly through hydrolysis.[Citation21]
As mentioned, hydrogels have the tendency to absorb large amount of water and accounts for approximately 10–20 times of their original molecular weight, when in fully swollen state and thus also referred to super absorbent hydrogels.[Citation22] Xerogels refers to the dried hydrogels, when the water molecules are removed from these swollen networks. As the removal of water from these swollen hydrogels cause no structural deformation, they are also termed as aerogels.[Citation23] The structure of the hydrogel network and the thermodynamic nature of the contents used in network development have a vital role in the diffusional performance, changes in the molecular mesh size and the related molecular stability of the built-in therapeutic agents.[Citation18] The swelling of the hydrogel network, drug loading, and release of the bioactive agents is directly related to the porous structure of the network. The porosity of the structure is mainly controlled by the density of the crosslinking agents in the gel matrix and the attraction of the hydrogels for the aqueous conditions in which the hydrogels becomes swollen. The porous structure of the hydrogels also allows the loading of the bioactive agents into the gel matrix and the succeeding release of the drug at a pre-determined rate which depends on the diffusion coefficient miniature molecules or large molecules contents through the hydrogel network.[Citation24]
The hydrogels network structure can be classified into nonporous, microporous, or macroporous depending upon the pore size. Nonporous hydrogels have a macromolecular aspect usually in the range of 10–100 μm, resembling mesh-like structures which are formed due to the crosslinking of monomer or polymer chains. The pore size of the microporous gel network is small commonly in the range of 100–1000 μm. The release of the therapeutic agents from this gel network occurs by diffusion and convection.[Citation23] The pore size of the macro porous hydrogels is usually of larger dimensions and ranges from 0.1 to 1 μm. These types of hydrogels release the bioactive agent that is entrapped inside the network pores through drug diffusion coefficient mechanism.[Citation25–27]
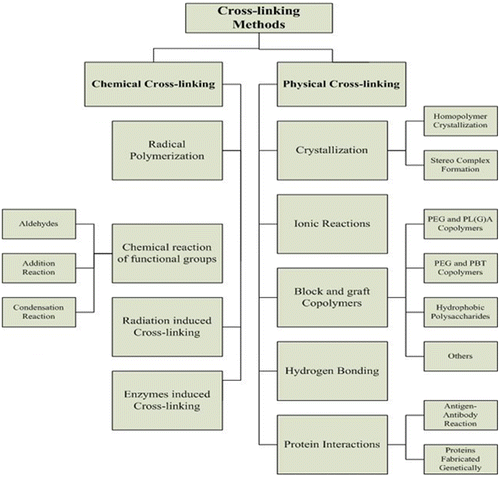
Hydrogels can be formulated either from natural polymers or synthetic sources. Numerous methods for the preparation of biomedical hydrogels have been reported in the literature. Hydrogels are either chemically crosslinked or have physical interaction between the polymer chains. Chemically crosslinked hydrogel formulations have covalent or ionic bonds between the polymer manacles or chains.[Citation28,29] Chemically crosslinked hydrogels can be prepared by either suspension polymerization, [Citation30] copolymerization, [Citation31,32] chemical reaction of corresponding functional groups, [Citation33–35] enzymatic crosslinking, [Citation36] polymerization through irradiation [Citation33]. For the preparation of chemically crosslinked hydrogels, crosslinking agents are used to develop crosslink’s between the polymer chains.
On the other hand, for the preparations of physically crosslinked hydrogels, several techniques have been implied such as hydrogen bonding, [Citation37,38] crystallization, [Citation39] protein interaction, [Citation40] ionic interaction, [Citation41–43] and by hydrophobic interaction.[Citation44,45]
Stimuli responsive hydrogels are nowadays gaining a wide interest in the field of drug delivery system and have numerous potential applications. These types of environment or stimuli-sensitive hydrogels are also called ‘smart’ or ‘Intelligent’ hydrogels. In the body, there are some environmental variables present such as high temperature and low pH. So, due to this reason, either temperature responsive or pH responsive hydrogels can be formulated and used for the controlled and site-specific drug delivery.[Citation46]
Hydrogels drug delivery has a wide range of applications in clinical practice and biomedical fields such as materials for contact lenses in tissue engineering, regenerative medicine, biosensors, and separation of biomolecules or matrices for cell-encapsulation and tools for the controlled and sustained release of bioactive agents and proteins due to their ability to fabricate in various physical forms.[Citation16,47–49] Drug delivery through hydrogels has gained a wide area of interest because of the ability to protect the bioactive agents from numerous hostile conditions such as low pH in the acidic environment of stomach and the presence of enzymes. The release of the therapeutic drug from the hydrogels drug delivery is mainly occurred at pre-determined rate and can also be controlled by modifying the gel network structure in reply to environmental stimuli e.g. temperature, pH, ionic strength, and electric field.[Citation50–53]
Calcium alginate microspheres were successfully used for the process of cell encapsulation by Lim and Sun in 1980.[Citation52] A new approach of designing was made by Yannas and co-workers in 1980s using hydrogels containing naturally occurring polymers like shark cartilage and collagen.[Citation54,55] For the process of cell encapsulation hydrogels of natural and synthetic source have been continuously used.[Citation56–59] In the field of tissue engineering, these hydrogels have been used for reforming various organs and tissues.[Citation60–67] Naturally, hydrogels are network of hydrophilic polymers which have the absorbing capacity ranging from 10 to 20% up to 1000 folds of their basic dry weight. Normally, hydrogels have shown chemical stability but they may dissociate by changing the environment. If ionic, hydrogen bonding or hydrophobic forces are involved in maintaining their molecular structure than they are called ‘physical hydrogels’ or reversible gels.[Citation68,69] Structure of hydrogel may not be uniform because they contain hydrophobic and ionic regions and clumps of molecules.
Ionotropic hydrogels are formed by the combination of polyelectrolyte with a multivalent ion. Calcium alginate is the most common example of ionotropic hydrogels. Gel precipitation may occur if polyelectrolytes of different charges are mixed depending on their PH, ionic strength, and concentrations. Complex coacervates and polyelectrolytes complexes are formed by crosslinking of ionic system. Calcium alginate capsules were stabilized using alginate poly (L-lysine) PPL by Lim and Sun [Citation54] in the field of tissue engineering polyion hydrogels and coacervates have been successfully used. Often, physical gels can be formed by using combination of polymeric biotin and avidin,[Citation70] or polymeric sugar and conavalin.[Citation71] The above-stated interaction could be reversed by changing temperature, pH, ionic strength, stress, and adding different solutes which have the competing capacity to displace ligands on the active site of protein. If hydrogels have covalent crosslinking, they are termed as chemical gels or permanent gels.
Wichterle and Lim prepared synthetic hydrogels by co-polymerizing EGDMA (crosslinker) with HEMA [Citation5]. If the nature of the polymer is changed from water soluble to water insoluble or molecular entanglement of hydrophilic polymers, it will produce chemical hydrogels. Sometime, crosslinking is not necessary in changing the hydrophobicity of a polymer to hydrophilicity. During the process of PAN hydrolysis, nitril group will dissociate into amide and an acid group. In the above reaction mixture, if the quantity of nitril group is enough, it will leads toward the stabilization of hydrogels, resulting in the formation of physical hydrogels. The optimum swelling ability of hydrogels relies on the concentration of crosslinking polymers. Chemicals hydrogels are not uniform as compared to physical hydrogels. Clumps having reduced water swelling capacity and enhanced crosslinking property are found in chemical hydrogels. High-density clumps are formed by the accumulation of water insoluble crosslinking polymers.[Citation72,73] During gel formation, different factors leads toward the phase separation or water filled spaces such as temperature, nature of solvent, and concentration of solids. Elasticity of chemical gels is not affected by the presence of free chain ends called defects.
Chemical and physical hydrogels can be prepared in different macromolecular forms. These forms are: complexes formed by polyion–polyion interaction. Various physical forms of hydrogels are solid molds, powders, microspheres, coated particles, thin layers, osmotic pumps, and liquids. Hydrogels are formulated by using a wide range of polymers having different composition as shown in Table . Hydrogels can be classified on the basis of composition of polymers which are natural, synthetic, and combination of these two. Figure shows different methods to prepare hydrogels.
Table 1. Water soluble polymers used for the preparation of hydrogels network.
Figure 1. Methods for the preparation of two different types of ionic hydrogels.
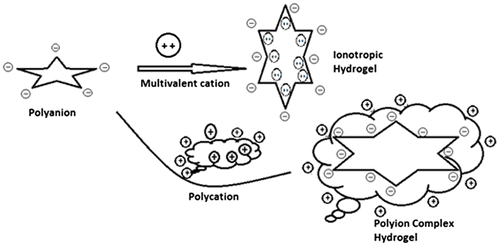
Figure 2. Represents methods for the preparation of hydrogels by chemical modification of hydrophobic polymers.
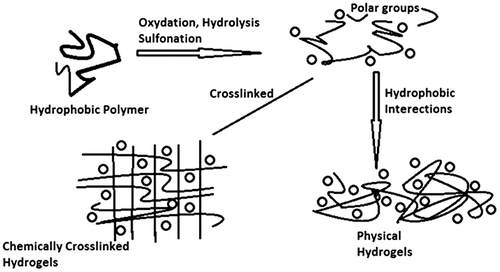
Figure 3. Represents methods used for the formulations of hydrogels by free radical reactions.
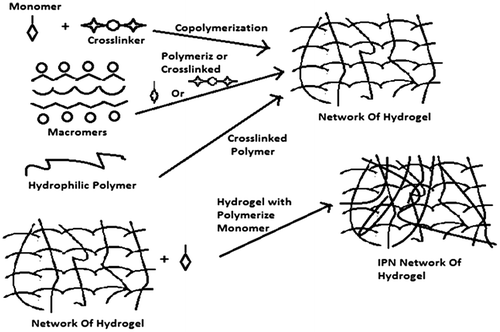
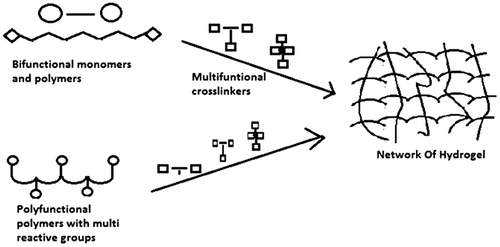
Figure 4. Represents methods used for the preparation of crosslinked hydrogels by condensation modes of reactions of polyfunctional reactants.
The aim of the current review is to report the ongoing research in the fields of hydrogels drug delivery system. In this review, we have discussed different methods used for hydrogels synthesis via graphical representations, classification of hydrogels. Channels or pores in hydrogel structure have been discussed in detail to understand the mechanism of permeation of fluid inside and outside of hydrogels network. Moreover, various methods of crosslinking used for the hydrogels, swelling characteristics of hydrogels, methods of drug loading, and drug release have been discussed in detail. This review highlights the synthesis and importance of different stimuli responsive/intelligent hydrogels such as pH, temperature, light, pressure, electro-sensitive, and glucose responsive. This review also includes the discussion of gel network engineering for optimum drug loading and release. The applications of hydrogels in pharmacy and medicine particularly in tissue engineering have been discussed.
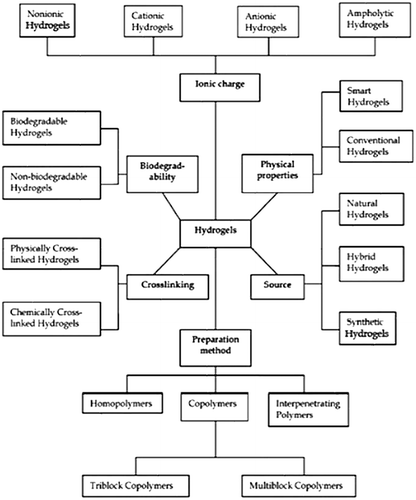
2. Classification of hydrogels
The classification of the hydrogels is mostly based on the presence of the nature of side functional groups in the polymer chains, which may be neutral or ionic. The swelling characteristics of the neutral hydrogels are mainly attributed to the overall unbound energy, imparted by water-polymer thermodynamic mixing along with expandable polymer contribution.[Citation18] Another classification of hydrogels is made on the basis of structural and mechanical characteristics which may be classified as phantom or affine. On the basis of methods of synthesis, hydrogels may be classified as copolymers or homopolymers. Moreover hydrogels can be classified on the basis of the physical characteristics of the hydrogels network structure such as amorphous, hydrogen bonded structures, semi-crystalline, super-molecular structures, semi-crystalline and hydro colloidal aggregates.[Citation74] Figure represents the classification of hydrogels.
Figure 5. Classification of hydrogels.
3. Structure of the hydrogel network
The appropriateness of a hydrogel as delivery tool for the bioactive agents and its role in a fastidious application largely depend on its bulk network structure. The important parameters used for the characterization of the hydrogels network structure include volume fraction of the polymer in the swollen state (V2s), molecular weight between the two adjacent nearest crosslinking points (Mc), and the consequent pore size of the network (€).[Citation75–79]
For the calculation of molecular weight between adjacent crosslink’s (Mc) of interpenetrating polymeric networks, Flory-Rehner theory is used. The (Mc) values represent the quantification of degree of crosslinking between the polymer chains, which may be either of physical nature or can be of chemical nature. So, because of the haphazard nature of the polymerization process, only average values of molecular weight between crosslinks (Mc) can be measured. Flory–Rehner equation used for the calculation of (Mc) values is given below.[Citation80]
(1)
Volume fraction of the polymer (V2s) in the swollen hydrogel is a measure of the quantity of fluid that a hydrogel can incorporate and contained into its structure. It is calculated by the following equation.[Citation81]
(2)
The porosity measurement or pore size of the hydrogels determines the porous structure of the hydrogels network. The pore size attributes to the correlation distance available between the polymer chains used for the diffusion of bioactive agents.
The release of many therapeutic agents from the hydrogels devices occurs primarily through the distances available between the polymer chains following the process of diffusion. This space or distances are considered as the ‘pore’. Hydrogels can be easily classified into many types depending on the size of the pores such as (1) microporous (2) macroporous and (3) nonporous. The pore size of the network structure can be calculated using the following equation.[Citation82]
(3)
3.1. Crosslinking and crosslink density
Crosslinker has an important role in altering the absorption and mechanical properties of hydrogels. Three-dimensional networks of polymers do not crosslink effectively at low concentration of crosslinking agent, whereas incorporation of higher percentage of crosslinking agent will involve large number of growing polymer chains to produce an additional network structure. As a result, swelling ratio and absorption capacity will decrease. Crosslinking density is controlled by the amount of crosslinking agent present in polymerization and double bond conversion.[Citation83] The crosslinking can be performed in two environments: in vitro during the preparation of a hydrogel or in vivo after application in a precise location of human body. Chemical crosslinking can be initiated by introducing a low-molecular-weight crosslinking agent together with a polymer into the reaction mixture.[Citation84]
4. Hydrophilicity in hydrogels
Transport of nutrients and cellular products through the hydrogel depends upon the role of water present inside the gel. As soon as absorption starts, the initial water molecule entering the gel will interact with the group having maximum polarity and hydrophilicity. This process is called primary attached water. After the hydration of polar networks, hydrogels start swelling and hydrophobic networks are exposed which then form bonds having hydrophobic nature and this is called secondary attached water. Combination of secondary and primary attached water is termed as total bond water. When polar and non-polar areas are completely hydrated, the gel will absorb extra water because of osmotic pressure. Elasticity in networking is produced by recantation force which opposes the extra swelling. In this way, hydrogel will attain its optimum swelling point. When all the polar, nonpolar, or ionic groups are completely saturated, the extra water (free water) is used to fill the empty spaces within the network. After the absorption of water, the network will swell up and the polymers will start to dissociate and dissolve which mainly depends on the nature and composition of the polymers. Special considerations are made for the hydrogels used in the field of tissue engineering i.e. they should not dry and the total water present should be calculated on the basis of free and bound water as mentioned earlier.[Citation3]
There are different methods to calculate the amount of bound and free water as part of total water present in the network. It has been proved through proton NMR that molecules interchange in every 9–10 s, so it is very difficult to precisely calculate the exact ratio of free and bound water. NMR, DSC, and molecular probes are the three main methods used for water characterization present in the gel. Labeled probe solution is used as standard in this process and the amount of probe molecules is measured when the process reach the equilibrium state. It is necessary that the free water molecule present in the hydrogel dissolve the probe solute molecules and thus we can measure the quantity of free water molecules by comparing the probe solute amount present in hydrogel and the amount present in external solution. The amount of water molecules present in bound form can be obtained by calculating the difference of total water content from free water content. It is also assumed that:
(a) Solute can be easily accessed by the free molecules of water present in the gel. (b) The quantity of solute in the external solution is equal to the quantity of solute in free water. (c) Networks in the gel are not affected by the quantity of solute. (d) The relative distribution of bound water and free water will not be affected by the amount of solute.
In DSC, it is assumed that the free water in the gel freezes, on heating, the freeze water will melt and will give the total amount of free water present. Amount of bound water is calculated by difference of free water and total water quantity.[Citation3,85] Table shows the different types of monomers and their abbreviations.
Table 2. Types of commonly used monomers and their abbreviations.
4.1. Channels and permeation in hydrogels
Permeation and diffusion of solute across the gel is calculated through total water content. During the preparation of gels, separation of phases leads to the formation of pores or they may already be present in the form of small pores within the gel network. The gel network is affected by various factors which are difficult to measure. These are distribution of pores, interaction between pores, and pore size. These factors are collectively called tortuosity of hydrogel.
Diffusion across hydrogel depends on the time duration, pore volume ratio, and thickness of the film. The formulation and density of the polymer will affect the above-stated factors. The different pore size in the hydrogel is produced by various molecular sizes and molecular weights of polymer probes.[Citation86] One of its examples is fluorescein-labeled dextrans. For the characterization of bound and free water, similar restrictions will be applied to the small molecular probes. One of the best and useful methods in order to characterize the pores in hydrogels and their interconnections is the probe solute permeation method in which solute permeation is governed by several important factors including probe hydrophilic and hydrophobic character, its size and shape, and the free water molecules availability to dissolve and hydrate the solute molecules.[Citation3]
For any specific hydrogel, the coefficient of permeation P is the product of apparent coefficient of diffusion (Dapp) and partition coefficient (K).The consistency of peptide and proteineceous drug loaded in hydrogel and partition coefficient K will rely on size, shape, and total charge size, total existing free water inside the hydrogel, the adding up of partition promoters to solution; pH, temperature ionic strength, and method of drying, if the hydrogel undergoes drying it often shades a high drug concentration at the hydrogel surface regions. If a proteineceous drug is loaded in a hydrogel, and the charge present on protein is opposite to that of hydrogel, then its pores might be plugged at surface while loading in gel and if its net charge is as same as that of hydrogel, then it might be expelled from the gel by Donnan exclusion process while loading protein in a gel, the pH and the buffer used in the protein solution and the ionic strength might separately or jointly control the quantity and supply of the protein loaded in the gel. The ‘apparent’ or ‘effective’ probe molecule diffusion coefficient (Dapp) is like to D0 times the proportion of the pore volume portion divided by tortuosity where D0 is the coefficient of diffusion in free water and ratio of pore volume portion divided by tortuosity is forever <1. Dissemination in alginate hydrogels has been studied in recent times.[Citation87] There will constantly be a section of the imbibed water in hydrogel which is not existing for drug permeation because of pore ‘dead ends’, little pores which are less than the diameter of drug molecule, hydrophobically or H-bonded ‘bound’ water, and drug matrix polymer relations. Release of high molecular weight drug from hydrogel will be restricted by pore volume portion, pore size, and their inter-connections, size of drug molecule, type and strength of interactions of drug with chains of polymer making up the network of hydrogel.[Citation88] In turn, key factors controlling the whole volume portion pore size and their inter-connections are the composition of network polymer chains and crosslink density. The connections of drug molecules with network chains will be resolute by their particular composition. So, in design, a hydrogel system for controlled drug release, it will be essential to ‘match’ the polymer make up and crosslink thickness with the particular size and composition of drug molecule to be delivered.
5. Methods of crosslinking to design hydrogels
Hydrogels have a wide range of applications in the delivery of bioactive molecules to different sites of the body. As the physiologic conditions of the body varies with different sites, these hydrogels drug delivery tools need stability in the varied physiological environment of the body. Since hydrogels are the crosslinked biomaterials, crosslinking in hydrogels chains provide stability to the network structure and retain them undissolved to maintain its three-dimensional structure in physiologic medium. In hydrogel formulations, various methods of crosslinking have been introduced. Generally, hydrogels are prepared by crosslinking the polymer chains either by chemical methods or physical means.[Citation89]
5.1. Chemically crosslinked hydrogels
Chemical crosslinking provides covalent bonds among the polymer chains which lead to the production of permanent hydrogel network.[Citation90] Chemically crosslinked gels are formed either by reactions between activated functional groups of polymer chains or by using small molecular weight crosslinkers, by triggering the reactions by photosensitive agents or by initiation with an enzyme.[Citation91,92]
5.1.1. Chemical crosslinking via condensation reactions
Polyesters and polyamides chains containing polymers are frequently obtained by condensation reactions of functional groups such as –OH or –NH2 with carboxylic acids or derivatives thereof.
Condensation reactions were used for the synthesis of hydrogels by most of the researchers in their work.
N, N-(3-dimethylaminopropyl)-N-ethyl carbodiimide (EDC), which is used for the crosslinking of water soluble polymers by creating amide bonds, is proven to be very efficient reagent. This reagent was used for the synthesis of gelatin hydrogels in their work by De Nooy et al. and Ichi et al. [Citation93,94] Tan et al. presented a method to prepare covalently crosslink alginate and PEG-diamines-based gels using EDC with better mechanical properties as compared to ionically crosslinked hydrogels.[Citation95] Passerini and Ugi condensation reactions were employed for the production of polysaccharide-based hydrogels by Lee et al. and Elbert et al. [Citation96,97].
5.1.2. Polymer–polymer crosslinking
Different researchers have suggested the use of activated reactive groups of pre-functionalized polymer chains which have reduced or completely eliminate the use of small molecule crosslinkers during hydrogel synthesis process. This strategy can also be used for the in situ synthesis of covalently bonded chemically crosslinked hydrogels. One of the examples is chitosan and Hyaluronic acid polymers based in situ hydrogels which were produced by Schiff bases formed at physiological pH. The gel was found stable for at least up to 4 weeks.[Citation98] Polymer–polymer linkages can also be formed by Michael addition reactions for chitosan hydrogels.[Citation99,100]
Hydrogels preparation by polymer–polymer interaction has many advantages but some disadvantages associated with their multi-step preparation and those concerned with purification process.
5.1.3. Chemical crosslinking via high energy irradiation
Permanent gels can also be produced through polymer crosslinking using high intensity and energy irradiations. Among these radiations, gamma and electron beams are most widely used. They are successfully used for the polymerization of hydrophilic polymers. For production of such hydrogels, vinyl group is polymerized to produce hydrogels.[Citation101] Hydrophilic polymers can also be crosslinked via high energy irradiations without presence of vinyl functional groups. Irradiation-induced chemical crosslinking involves the production of free radicals on the hydrophilic polymer chains.[Citation102] These chains are involved in polymerization process. During polymerization, hydroxyl radicals attack the polymer chain and produce macro radicals. These macro radicals then synthesize covalent bond between polymer chains followed by the formation of crosslinked networks.[Citation74] Some of the polymers which are generally crosslinked via high energy irradiation includes (acrylic acid) [Citation103] poly (ethylene glycol) [Citation104–106] and poly (vinyl alcohol).[Citation107] The fate of hydrogel properties such as swelling ratio and permeability mainly depends upon the contents of polymers used and irradiation dose.[Citation108,109] Under mild conditions of pH and temperature, hydrogel can be synthesized by high energy radiations mediated crosslinking keeping in mind the avoidance of usage of toxic crosslinking agents.
5.1.4. Crosslinking using enzymes
Nowadays, enzymes are also used for polymer chains crosslinking to produce covalently crosslinked hydrogels. PEG-based hydrogels are the most common example produced enzymatically. In this study, a tetrahydroxy PEG was modified with glutaminyl groups (PEG-Q). Trans-glutaminase was added to aqueous solutions of poly (lysine-co-phenylalanine) and PEG-Q to produce PEG networks. This enzyme induce catalytic reaction between the carboxamide group and the amine group of lysine, resulting in amide linkage between the polymers.[Citation110] Recently, Sperinde et al. synthesized poly (lysine-co-phenyl-alanine) by lysine end-functionalized PEG. In this work, aqueous solution of peptide-modified macromers was prepared and transglutaminase was added to it. It was found that the kinetics of gelation mainly depends upon concentration of enzyme, the structure and composition of macromer and ratio of the reactants.[Citation111]
5.2. Physically crosslinked hydrogels
In the recent past, physically crosslinked hydrogels have gained huge interest. One of the advantages of such type of hydrogels is the elimination of crosslinking agent used in chemically cross-linked hydrogels. It is also reported that some crosslinking agents have toxicity problems, so their elimination from the gel network prior to application is necessary. Reported methods for the preparation of physically crosslinked hydrogels includes;
5.2.1. Charge interactions
The phenomena of charge interactions between two or more oppositively charged polymers have been widely explored for the production of physically crosslinked in situ gelling polymers. Hydrogel may be formed by interaction between two oppositively charged polymers or a polymer and another small molecule. Example of the physically crosslinked hydrogels by this method is the crosslinking of elastin like polypeptides by electrostatic interactions under specified physiological conditions between cationic lysine residues and anionic organophosphorus crosslinker.[Citation112]
In this approach, the crosslinked network is broken down by ionic species mainly present in the extracellular fluid that are bound to gelling components. Crosslinking of microgels and nanogels can be performed using charge interaction approach to deliver therapeutic agents and some macromolecules. Example includes dextran microspheres, these spheres were coated with oppositively charged polymers. The resulted microsphere showed spontaneous gelation due to formation of ionic complex.[Citation113]
5.2.2. Crosslinking by crystallization
Physically crosslinked hydrogels can also be produced by crystallization approach. The example is the physically crosslinked poly (vinyl alcohol) (PVA) hydrogels. PVA is a water soluble polymer and its aqueous solution slowly forms a gel at room temperature. Mechanical strength of these hydrogels was low, but after freeze-thaw treatment of aqueous solution of polymer, a strong and highly elastic gel was formed.[Citation114] Major factors that influence the properties of the formed gel includes; PVA molecular weight, its concentration in water, the number and time duration of freeze-thaw cycles and the temperature. Gel that was formed was due to the formation of PVA crystallites. These crystallites act as physical crosslinking sites in the hydrogel network. Report revealed that this hydrogel was stable at 37 °C for 6 months and formation of PVA gels by crystallization approach is also known as hydrogen bonding interactions.
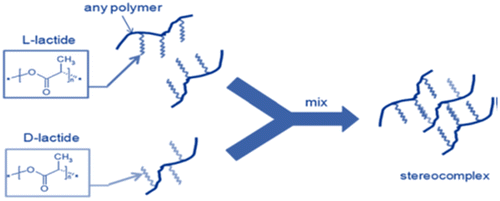
5.2.3. Crosslinking by stereocomplex formation
In recent past, hydrogels are also prepared on the basis of stereocomplex formation. This approach is based upon the synergistic interactions between chains of polymer or small molecules having different stereochemistry but same chemical composition.[Citation115] Example is the homopolymers of Poly (l-Lactic acid) and Poly (d-Lactic acid), both of these are semi-crystalline materials. Interaction between these homopolymers is due to the stereocomplex formation.[Citation116] Stereocomplex grafting approach can also be used for crosslinking of natural polymers. Dextran precursors are grafted to the l, d-Lactide oligomers which results in spontaneous gelation in water. The process is carried out without the use of denaturing agents, for example, organic solvents, crosslinking agents, and formation of hydrophobic domains. Gels produced by this approach have excellent biodegradability and biocompatibility. One of the limitations of this crosslinking approach is the use of specified number and range of polymers. Figure indicates the stereocomplex method used for crosslinking of hydrogels.
Figure 6. Crosslinking of hydrogels by stereocomplex formation.
6. Stimuli-responsive hydrogels
Stimuli-responsive or so-called ‘smart’ polymers have gained great attention for their use in drug delivery approaches in the recent past. These polymers respond to physical or chemical stimuli such as change in pH, temperature, ionic strength, light, the presence of enzymes or a specific ligand, and electric or magnetic fields.[Citation117] Some polymer systems respond to more than one stimuli, for example, the response of a polymer to both pH and temperature.[Citation118,119]
Temperature and pH responsive polymers are more suitable and feasible candidates for drug delivery systems because they undergo conformational changes due to change in external environment. These changes can be effectively used to enhance drug loading, site of the drug delivery, and rate of the drug release.
Environmentally sensitive hydrogels have the ability to sense changes of pH, temperature, ions, light, or the concentration of metabolite. Natural hydrogel materials are being investigated for tissue engineering, which include agarose, methylcellulose, and hylaronan. Figure represents the different methods of crosslinking. Some of the environmental responsive hydrogels system include;
Figure 7. Methods of crosslinking of hydrogels.
6.1. Temperature-sensitive hydrogels
Temperature responsive polymers are the most widely investigated in modern drug delivery approaches. They have the characteristic to change their conformation, hydrophilic/lipophilic balance, and solubility as a response to temperature change in the external environment.[Citation117,120] The components of polymers are insoluble above or below a specific temperature termed as the upper and lower critical solution temperature (UCST and LCST). Polymers with LCST are more commonly used in drug delivery systems as compared to the polymers with UCST. The interaction forces i.e. hydrogen bonding between water molecules and polymer weakens at LCST as compared to polymer–polymer and water–water interaction. This weakness of interaction leads to phase separation with ultimate result of gel formation.[Citation121] The gels are formed by physical reversible linkage in the chains of polymers due to temperature stimulus. By removing the stimulus, the gels can return to its solution form due to decrease in hydrophobicity. At ambient temperature, these solutions have low viscosity and are free flowing but at body temperature, they form a semi-solid gel.[Citation122] Commonly used thermos-responsive polymers for controlled or sustained drug delivery include; N-isopropylacrylamide, polysaccharides (e.g. chitosan, cellulose derivatives, xyloglycan,), and Pluronics (e.g. poloxamers, (PEG-PLA-PEG) and N-vinylcaprolactam.[Citation123]
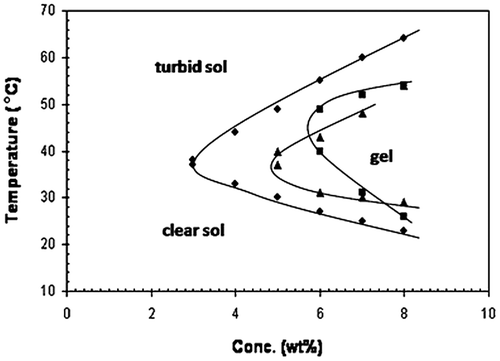
In controlled drug delivery, the most important concept is in situ gelling systems. A low viscosity solution can be prepared using thermo-responsive polymers. This solution has the advantage of easy injection and undergoing fast gelation in situ in response to body temperature. Figure shows the sol–gel conversion of thermosensitive system. Figure represents the effect of temperature on drug release from thermoresponsive hydrogel system. Figure represents the effect of concentration and temperature on sol–gel conversion.
Figure 8. The sol–gel conversion of thermo-sensitive system.
Figure 9. Effect of temperature on drug release from a thermo-sensitive hydrogel.
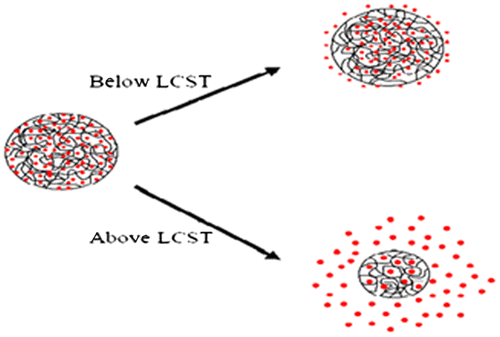
Figure 10. Effect of concentration and temperature on sol–gel conversion.
6.2. pH-responsive hydrogels
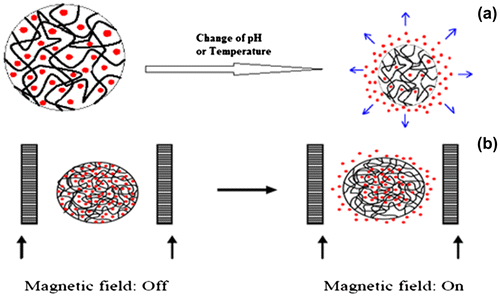
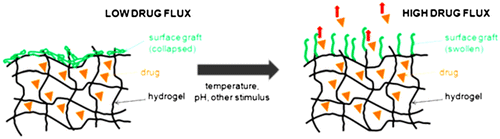
Polymers that respond to pH typically contain ionizable groups that can accept or donate protons in response to the environmental pH. In this case, changing the pH across the pKa threshold of the polymer results in a rapid change of its net charge and in the hydrodynamic volume of the polymer chains. In other words, the presence of a highly charged structure causes the matrix to swell as a result of the high charge repulsion, and as the polymer shifts from a collapsed state to an expanded state.[Citation124] As mobile counter ions are present in the polymeric network which creates osmotic pressure. Increase in osmotic pressure thus explains the increase in the hydrodynamic volume of the polymer chains.[Citation125]
There is remarkable pH difference in the human body particularly along the gastrointestinal tract. This pH difference can be exploited for targeted drug delivery using pH responsive polymers. Extracellular pH of the tumors is acidic which helps in enhancing the release of anti-cancer drug from a pH responsive polymer, increasing the efficiency and decreasing target to non-target ratio.[Citation126,127]. pH responsive polymers include; Polybases such as poly (N, N′-dimethyl aminoethyl methacrylate) (PDMAEMA) and poly (amino esters). These polymers have amino groups and at neutral pH, they are deprotonated, but at acidic pH, they gain protons and become positively charged.[Citation124] This is particularly relevant in cancer drug delivery, since the extracellular environment of solid tumors is weakly acidic. Figure represents the effect of different environment on hydrogels.
Figure 11. Environmentally responsive drug delivery systems: (a) pH and temperature sensitive and (b) magnetic sensitive.
6.3. Glucose-responsive hydrogels
Insulin delivery in a self-regulated mode is among the major problems in the area of controlled drug delivery. Glucose responsive polymers can be used to overcome this problem. These polymers have the ability to initiate secretion of endogenous insulin hence minimizes diabetic complications by releasing the bioactive compound in a sustained manner. These polymers are sensitive to sugar and show variable response to the presence of glucose. They have earned remarkable attention due to their use in both insulin delivery as well as glucose sensing. There is major difference between delivery of insulin and other drugs because insulin is delivered in specific amount at specified time when needed. Keeping in mind these parameters and limitations, insulin delivery systems are designed in such a way that it has the ability to sense glucose and automatic shut-off mechanism when there is no need of insulin. All the hydrogel systems developed for insulin delivery have a glucose sensor. Enzymatic oxidation of glucose results in production of byproducts. Polymers used in such a system are sensitive to these byproducts and, hence, show response to glucose. Glucose is oxidized by glucose oxidase and form gluconic acid and H2O2.[Citation128]
These have many advantages but the most common problems are its short response time and the possibility of non-biocompatibility. Basic approach for the development of glucose-responsive polymeric systems is: oxidation of glucose by glucose oxidase, and glucose binding with lectin or formation of reversible covalent bond with phenyl buronic acid moieties.[Citation129]
6.4. Electro-sensitive hydrogels
Some polymers change their physical properties in response to minor changes in electric field sensing it as environmental stimuli.
Hydrogels sensitive to electric current are usually made of polyelectrolytes, as are pH-sensitive hydrogels. Electro-sensitive hydrogels undergo shrinking or swelling in the presence of an applied electric field.[Citation130] It is also possible, that sometimes the hydrogels show swelling on one side and deswelling on the other side and results in bending of the hydrogels. This happens due to change in pH caused by electric current. pH change disrupts the hydrogen bonding between chains of the polymer resulting in degradation or bending of the polymer chain and ultimately release the drug. Mechanisms of drug release involve diffusion, charged drug electro-phoresis, and forced release of the drug from electro-erodible polymers upon its erosion. Most commonly used natural polymers for preparation of electro-responsive hydrogels include; hyalouronic acid, chitosan and alginate, while synthetic polymers include; vinyl alcohol, methacrylic acid, allyl amine, vinylacrylic acid, and acrylonitrile.
Electric energy is transformed into mechanical energy in electro-responsive polymers. They have been widely used in the field of artificial muscle activation and propulsion, controlled drug delivery, sound suppression, and energy transductions Neutral polymers that exhibit electro-sensitive behavior require the presence of a polarizable component with the ability to respond to the electric field. A rapid bending of gel in silicon oil was observed in the case of lightly crosslinked poly (dimethyl siloxane)-containing electro-sensitive colloidal SiO2 particles.[Citation131]
Electro-responsive polymers transform electric energy into mechanical energy and they have wide application in the field of controlled drug delivery, artificial muscle actuations, and energy transductions, and sound dampening.
Electro-sensitive hydrogels have been applied in controlled drug delivery.[Citation132] Hydrogels prepared from poly (2-acrylamido-2-methylpropane sulfonic acid–co-n-butyl methacrylate) released edrophonium chloride and hydrocortisone in a sustained manner by applying electric current.[Citation133] Control of ‘on–off’ drug release was achieved by varying the intensity of electric stimulation in distilled–deionized water. For edrophonium, a positively charged drug, the release pattern was explained as an ion exchange between the positively charged solute and hydrogen ion produced by electrolysis of water.
6.5. Light-sensitive hydrogels
These hydrogels are most widely used in the development of display units, optical switches, and ophthalmic drug delivery devices. Light-sensitive hydrogels have many advantages as compared to others because light stimulus can be instantly applied in specific amounts with high accuracy. For example, rate limiting step for pH sensitive hydrogels is hydrogen ion diffusion, while for temperature-responsive hydrogels, the rate limiting step is the thermal diffusion.
Light-sensitive hydrogels can be categorized into visible light-sensitive and UV-sensitive hydrogels. As compared to UV light, visible light is safe, inexpensive, easily available, clean, and conveniently controlled.
The UV-sensitive hydrogels were synthesized by introducing a leuco-derivative molecule, bis (4-dimethylamino) phenylmethyl leucocyanide, into the polymer network.[Citation134] Triphenyl methane leuco-derivatives are neutral under normal conditions but upon exposure to ultraviolet irradiation, it dissociate into ion pairs and produce triphenyl-methyl cations. When UV irradiation was applied at fixed temperature, the hydrogels swelled discontinuously, followed by shrinkage upon removing the UV light. There is difference between discontinuous and continuous volume phase transition induced in the presence and absence of UV light, respectively. UV irradiation produces cyanide ions which increases the osmotic pressure within the gel results in swelling of gel.
These hydrogels have application in the development of memory devices, photo-responsive artificial muscles, and switches.[Citation135] Visible light-responsive hydrogels were also proposed for their potential use in temporal drug delivery, based on crosslinked hyaluronic acid hydrogels response to the light. These hydrogels undergo light-mediated degradation in the presence of methylene blue.
6.6. Pressure-sensitive hydrogels
Thermodynamic calculations based on uncharged hydrogel theory led the foundation of the concept that pressure may induce volume phase transition in hydrogels. This theory claims that the same hydrogels collapsed at low pressure will expand with increase in pressure. This prediction was confirmed by experiments with poly (N-isopropylacylamide) hydrogels.[Citation136] Under condition of hydrostatic pressure and temperature near LCST, the degree of swelling of poly (N-isopropylacylamide) hydrogels increases remarkably. Some other hydrogels e.g. poly (N, N-diethyl acrylamide), poly (N-isopropylacylamide), and poly (N-n-propylacrylamide) showed sensitivity to pressure at temperature near their LCSTs. Sensitivity to pressure appears to be a common feature of thermo-sensitive gels. It was concluded that the LCST value of thermo-sensitive gels increase with increase in pressure and thus show response to pressure.[Citation137]
6.7. Specific antigen-responsive hydrogels
Development of a device or material sensitive to specific proteins is of prime importance in biomedical applications. On the basis antigen-antibody interaction, sol–gel hydrogels were prepared having phase reversing property. These hydrogels are based on the same concept used in glucose-responsive phase-reversible hydrogels. During preparation of these semi-interpenetrating hydrogels, a specific antigen and its antibody were grafted into the network of different polymers.[Citation138] Binding of an antigen and antibody results in crosslinking interactions and forms a gel. Hydrogel swelling is enhanced when free antigens are present that compete with antigens bound to the polymer and thus crosslinking density is reduced.
6.8. Thrombin-induced infection-responsive hydrogels
Grafted gentamycin was loaded into PVA hydrogels to release the drug at the specific site of infection. During preparation, gentamycin was attached chemically to the backbone of the polymer using peptide linkers that are prone to enzymatic degradation e.g. degradation by thrombin.[Citation139] A high thrombin-like activity toward specific peptide sequence was observed from dorsal pouch exudates of Pseudomonas aeruginosa-infected rats as compared to the exudates from non-infected wounds. This approach is same as the slow drug release attached to polymeric prodrugs that release the attached drug molecules slowly but the difference is that the release is triggered by infection.
This type of approach can be applied to occlusive wound dressings and infection-prone catheters, drainage bags, and prostheses.[Citation140] This system comparatively have high specificity and good potential as a stimuli-responsive, controlled release drug delivery system.[Citation141]
7. Gel network engineering
Several attempts have been made to modify the microstructure of the hydrogel to control the drug diffusion from the gel matrices. Gel microstructure is modified either at the surface (locally) or throughout the gel network. This modification can be simply performed by increasing the percentage of crosslinker keeping in mind the limitations because high crosslinking results in slow response of the gel to the external stimuli and may also induce certain unwanted mechanical properties.
7.1. Interpenetrating polymer networks
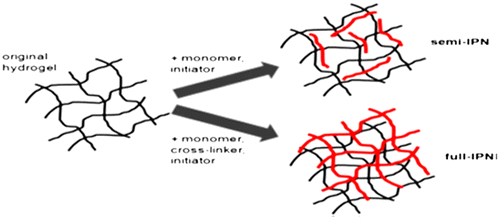
Polymerization of a pre-polymerized hydrogel by another hydrogel network leads to the formation of an interpenetrating polymer network (IPN). During this process, a pre-polymerized hydrogel is immersed into a solution containing monomers and an initiator. IPNs are formed both in the presence and absence of a crosslinker. In the presence of a crosslinker, a fully interpenetrating polymer network (full IPN) is formed, while in its absence, a network is generated in which embedded linear polymers are entrapped within the network of the original hydrogel (semi-IPN), as shown in Figure . IPNs have many advantages as dense hydrogels can be produced which are comparatively stiffer and tougher, their physical properties can be controlled more widely and also have efficient drug loading as compared to the conventional hydrogels. Drug loading and polymerization are performed simultaneously.[Citation142]
Figure 12. Represents the formation and structure of semi and full IPNs.
Modifications in pore size and surface chemistry also affect the tuning of mechanical properties, hydrogel-tissue interaction, and drug release kinetics.[Citation143]
Semi-IPNs can more effectively preserve rapid kinetic response rates to pH or temperature (due to the absence of a restricting interpenetrating elastic network) while still providing most of the benefits of IPNs in drug delivery (e.g. modifying pore size, slowing drug release, etc.). For example, entrapping linear cationic polyallylammonium chloride in an acrylamide/acrylic acid copolymer hydrogel imparted both higher mechanical strength and fully reversible pH switching of theophylline release.[Citation144]
7.2. Surface diffusion control
Surface can be modified as an alternative to bulk structure modification of a hydrogel. In such modifications, a thin layer ‘film’ is generated at the surface of hydrogel having reduced permeability and thermo-responsive on–off switch mechanism of drug release. Control of drug diffusion through this approach is shown in Figure . Using this approach, a temperature-dependent surface permeability and drug release kinetics can be achieved by grafting thermo-sensitive polymers on to the hydrogel surface such as PNIPAM polymers.[Citation145] At low temperature, the drug is released rapidly but at higher temperature, it is remarkably slowed because at higher temperatures, there is phase transition and the thermos-responsive polymer collapses onto the surface of the hydrogel. Alternately, drug diffusion from the bulk hydrogel can be controlled by coating with a multilayer dense film of polyelectrolyte,[Citation146] limiting drug diffusion out of the bulk hydrogel. The diffusion rate is designed in such a way that it is either dependent on the pH of the medium used or rate of degradation of the film. It may also be dependent on the environmentally dependent swelled form of the coated hydrogel. Film is ruptured due to exertion of mechanical pressure that is exerted on the coating causing burst release of the drug at specified conditions.[Citation147]
Figure 13. Shows the drug diffusion control by surface-modifying a hydrogel with an environmentally responsive polymer graft.
8. Hydrogels as sensors
Hydrogels are hydrophilic, highly water swellable polymer networks capable of converting chemical energy into mechanical energy and vice versa. Over the last decade, these materials have gained considerable recognition as valuable tool for sensors and in diagnostics.
In general, sensors are defined as
A sensor is a device that receives and responds to signals and stimuli from the environment.
In nature, human being gave the first idea of sensors such as sight, smell, touch, taste, and hearing which includes some of the physical and chemical stimuli.
In 1991, IUPAC classified sensors as physical and chemical. Physical sensors refers to a device which gives information about the physical nature of a system
While chemical sensor tells us about the chemical information, which ranges from the concentration of a specific sample component to total composition analysis, into an analytically useful signal.
In 1999, IUPAC defines the term biosensors which is defined as
A biosensor is an integrated receptor-transducer device, which is capable of providing selective quantitative or semi-quantitative analytical information using a biological recognition element.
Biosensors are becoming increasingly important and practical tools to suit a vast pool of application areas including point-of-care testing, home diagnostics, and environmental monitoring.[Citation148]
Hydrogels may be prepared in aqueous solutions by UV [Citation149] or thermo-initiated radical polymerization,[Citation150] addition reaction,[Citation151] self-assembly of recognition motifs such as coiled-coils,[Citation152,153] peptides,[Citation154–156] hydrogen-bridges, [Citation157] or DNA.[Citation158,159] These methods can be used in a huge number of particular techniques for the manufacture of strange hydrogel assemblies that are particularly useful for sensing.
Stimulus-sensitive hydrogels may perform as active sensing material. Such gels show sensitivity to little changes in the environment and give the response to physical stimuli (temperature,[Citation160] light, pressure,[Citation161] electric field,[Citation162] ionic strength,[Citation163] and magnetic field [Citation164]), chemical stimuli (pH,[Citation165] ions [Citation166] or biological stimuli (glucose [Citation167], enzyme, [Citation168] and antigen [Citation169]) through volume changes. In the presence of these stimuli, the hydrogels undergo phase transition by the sensing molecules and simultaneously translate this sense into a macroscopic event.
A direct application of hydrogels in biosensors is a safeguarding and coating function of sensor parts to avoid the unwanted interaction with biological molecules or cells.
Hydrogels can also be used as immobilization matrix for the biosensing elements. In designing biosensors, the essential criteria with high selectivity and sensitivity are the receptors that have to be immobilized so that could be able to specifically interact with the analyte, and the support material needs to show resistant to unspecific adsorption.
In hydrogel-based sensors, recognition between an analyte and sensing element occurs by volume changes in response to target molecules. This recognition made volumetric change generates a new type of sensing system as alternative to typical biosensors based on electrochemical sensing. Figure shows the types of biosensors.
Figure 14. Types and components of biosensors as well as their fields of application. Figure redrawn from [Citation110].
![Figure 14. Types and components of biosensors as well as their fields of application. Figure redrawn from [Citation110].](/cms/asset/788d7bec-03d6-4b77-8726-077b6c2378a0/tdmp_a_1169380_f0014_oc.gif)
9. Drug loading and release
The physical and chemical properties of the hydrogels are very important in the delivery of therapeutic substances. The three main methods used for the loading of bioactive agents in hydrogels include entrapment, tethering, and diffusion. Depending on the network and nature of therapeutic substance, a method is selected which has specific advantages and disadvantages.[Citation170–172]
The simplest method of drug loading is the diffusion method which includes placement of hydrogel system in the solution containing a therapeutic agent. Depending upon the hydrogel network, size of the drug molecules and chemical properties, the payload diffuses slowly into the network.[Citation173]
In case of proteins, peptides and larger drug molecules, which cannot be diffused through the small pores, the payloads are entrapped during the gelation process. The processes of diffusion and entrapment allow the free passage of the payloads in and out of the gel network. But these approaches offer some sort of toxicity by the initial burst release of the drug by invivo placement of the hydrogels system.[Citation174]
In order to avoid this risk, the payloads are attached to the network structure through physical or covalent linkages. This tethering method limits the exposure of the tissue to the agent, when it is broken.[Citation175]
Release of the active agents from the gel network occurs through enzymatic specific stimulation, environment specific stimulation, or diffusion. Diffusion involves the movement of molecules through the network or bulk erosion of the gels. Environmental sensitive hydrogels show response to some physical stimuli and release the drug in response to that stimuli. The new release mechanism involves the release of payload by local enzymatic activation and degradation. These biochemically stimulated responses occur by tethering drugs to the hydrogel via labile domains that are susceptible to matrix remodeling enzymes or using polymers that are targeted by enzymes.[Citation176,177]
10. Application of hydrogels as tissue engineering matrices
When part or the entire of definite tissues or organs stop working, there are numerous options for management including renovation, substitution with artificial or innate substitute, or rejuvenation. Tissue renovation or substitution with an artificial alternate is restricted to those situations wherever surgical method and implant have gained success.
Tissue engineering has immense pledge for rejuvenation of organs. Hydrogels have been progressively more studied as matrices regarding tissue engineering.[Citation178] Hydrogels made to be utilized as scaffolds in tissue engineering might contain pore huge sufficient to lodge alive cells, or may be structured to liquefy or degrade away by releasing growth factor and creating pore in which alive cells might go through and reproduce. Table is identifying the significant advantages and shortcomings of hydrogels as the matrices of tissue engineering. One important benefit of hydrogels as tissue engineering matrices vs. extra hydrophobic alternative like PLGA is the easiness because of which one might covalently slot in cell membrane receptor peptide ligand, in order to inspire adhesion, scattering, and growth of the cells in the hydrogels matrix.
Table 3. Merits and demerits of hydrogels as matrices in tissue engineering.
However, an important shortcoming of hydrogels is their little mechanical strength, posturing important difficulty in handling.[Citation179] Sterilization issue is also too much challenging. It is evident that there are both important advantages and disadvantages regarding use of hydrogels in tissue engineering, and the final will require being triumph over before hydrogels will become realistic and valuable in this electrifying field. Table indicates the examples of some commercially available hydrogel-based products.
Table 4. Examples of commercial hydrogel-based products.
11. Summary and future prospective
Hydrogels due to their tremendous diversity and intrinsic properties have gained extensive interest of the scientists and determine applications in pharmaceutical and biomedical fields. Stimuli-responsive hydrogels have great potential in the delivery of pharmaceuticals due to their response to various stimuli such as low pH and elevated temperatures found in the body. This property of hydrogels enables functionalization of hydrogels to be introduced with such moieties that make them biosensors. Hydrogels may be prepared through a variety of methods including physical and chemical crosslinking methods most notably via radical polymerization technique. Moreover, hydrogels have the ability to be loaded with different active agents having variable solubility, genes, enzymes, antibodies, biochemical recognition sites, dyes, etc. due to their porous structure. The loading of bioactive agents and other pharmaceutical and biotechnological products can be achieved by a variety of methods such as diffusion, tethering, and entrapment. Release of the bioactive agents can occur via enzyme specific stimulation, diffusion, and environment-specific stimulation. Nowadays, insitu hydrogels are gaining increased interest which are in solution state at room temperature and converts into gel form at physiological temperature. These insitu hydrogels act as depot system for local and systemic delivery of pharmaceuticals. Hydrogels network may be semi-interpenetrating (semi IPN) or full interpenetrating (full IPN) depending upon the absence or presence of crosslinker. Hydrogels can be used as biosensors in which it can be used as immobilization matrix for biosensing elements. Hydrogels have a wide range of applications in pharmaceutical and medical field. In the field of tissue engineering, hydrogels have been extensively studied as tissue matrices or scaffolds. Future development of the hydrogels may include the combination of modern biotechnological techniques for preparation and designing hybrid hydrogels system which may prove an asset in the field of drug delivery.
Funding
This work was supported by COMSATS Institute of Information Technology.
Disclosure statement
No potential conflict of interest was reported by the authors.
Acknowledgments
The authors thank Dr Nisar-ur-rehman, Head of the Department of Pharmacy, COMSATS Institute of Information Technology, Abbottabad Pakistan for his valuable guidelines.
References
- Bajpai SK, Saggu SS. Insulin release behavior of poly (methacrylamide-co-N-vinyl-2-pyrrolidone-co-itaconic acid) hydrogel: an interesting probe. Part II. J. Macromol. Sci. A: Pure. Appl. Chem. 2007; 44:153–157.
- McNeill ME, Graham NB. Properties controlling the diffusion and release of water soluble solutes from poly (ethyl oxide) hydrogels 1. J. Biomater. Sci. Polym. 1993;4:305–322.10.1163/156856293X00582
- Hoffman AS. Hydrogels for biomedical applications. Adv. Drug Deliv. Rev. 2002;54:3–12.10.1016/S0169-409X(01)00239-3
- Peppas NA, Hilt JZ, Khadem hosseini A, et al. Hydrogels in biology and medicine: from molecular principles to bionanotechnology. Adv. Mater. 2006;18:1345–1360.10.1002/(ISSN)1521-4095
- Wichterle O, Lim D. Hydrophilic gels in biologic use. Nature. 1960;185:117.10.1038/185117a0
- Hoffman AS, Schmer G, Harris C, et al. Covalent binding of biomolecules to radiation-grafted hydrogels on inert polymer surfaces. Trans. Am. Soc. Artif. Intern. Organs. 1972;18:10–16.10.1097/00002480-197201000-00003
- Ratner BD, Hoffman AS. Synthetic hydrogels for biomedical applications. In: Hydrogels for medical and related applications. ACS symposium series, Vol. 31. Washington, DC: American Chemical Society; 1976. p. 1–36.10.1021/symposium
- Peppas NA, editor. Hydrogels in medicine and pharmacy, Vols. I–III. Boca Raton, FL: CRC Press; 1987.
- Park K, Shalaby WSW, Park H, editors. Biodegradable hydrogels for drug delivery. Lancaster, PA: Technomic; 1993.
- Harland RS, Prud’homme RK, editors. Polyelectrolyte gels: properties, preparation, and applications. Washington, DC: American Chemical Society; 1992.10.1021/symposium
- Ulbrich K, Subr V, Podperová P, et al. Synthesis of novel hydrolytically degradable hydrogels for controlled drug release. J. Controlled Release. 1995;34:155–165.10.1016/0168-3659(95)00004-R
- Hoffman AS. Intelligent polymers. In: K Park, editor. Controlled drug delivery. Washington, DC: American Chemical Society; 1997.
- Harris JM, Zalipsky S, editors. Poly (ethylene glycol) chemistry and biological applications, ACS Symposium Series. Washington, DC: American Chemical Society; 1997.10.1021/symposium
- Baroli B. Hydrogels for tissue engineering and delivery of tissue-inducing substances. J. Pharm. Sci. 2007;96:2197–2223.10.1002/jps.20873
- Entezami AA, Massoumi B. Artificial muscles, biosensors and drug delivery systems based on conducting polymers: a review. Iran. Polym. J. 2006;15:13–30.
- Ghazizadeh Y, Mirzadeh H, Amanpour S, et al. Investigation of effectiveness of chitosan hydrogel to stop bleeding and air leakage from lung fistula: an in vivo study. Iran. Polym. J. 2006;15:821–828.
- Gao D, Xu H, Philbert MA, et al. Ultrafine hydrogel nanoparticles: synthetic approach and therapeutic application in living cells. Angew. Chem. Int. Ed. 2007;46:2224–2227.10.1002/(ISSN)1521-3773
- Peppas NA, Buresa P, Leobandunga W, et al. Hydrogels in pharmaceutical formulations. Eur. J. Pharm. Biopharm. 2000;50:27–46.10.1016/S0939-6411(00)00090-4
- Peppas NA, editor. Hydrogels in medicine and pharmacy, Vol. I, II, III. Boca Raton, FL: CRC Press; 1986.
- Kamath K, Park K. Biodegradable hydrogels in drug delivery. Adv. Drug Deliv. Rev. 1993;11:59–84.10.1016/0169-409X(93)90027-2
- Park K, Shalaby WSW, Park H, editors. Biodegradable hydrogels for drug delivery. Basle: Technomic; 1993.
- Kim SW, Bae YH, Okano T. Hydrogels: swelling, drug loading and release. Pharm. Res. 1992;9:283–290.10.1023/A:1015887213431
- Amin S, Rajabnezhad S, Kohli K. Hydrogels as potential drug delivery systems. Sci. Res. Essay. 2009;3:175–1183.
- Sutton C. Adhesions and their prevention. Obstet. Gynaecol. 2005;7:168–176.10.1576/toag.2005.7.3.168
- Aroca AS, Ribelles JL, Pradas MM, et al. Characterization of macroporous polymethyl methacrylate coated with plasma polymerized poly 2-hydroxyethyl acrylate. Eur. Polym. J. 2007;43:4552–4564.10.1016/j.eurpolymj.2007.07.026
- Rowley J, Madlambayan G, Faulkner J, et al. Alginate hydrogels as synthetic extracellular matrix materials. Biomaterials. 1999;20:45–53.10.1016/S0142-9612(98)00107-0
- Liu Q, Hedberg EL, Liu Z, et al. Preparation of macroporous poly 2-hydroxyethylmethacrylate hydrogels by enhanced phase separation. Biomaterials. 2000;21:2163–2169.10.1016/S0142-9612(00)00137-X
- Satish CS, Satish KP, Shivakumar HG. Hydrogels as controlled drug delivery systems: synthesis, cross-linking, water and drug transport mechanism. Ind. J. Pharm. Sci. 2006;68:133–140.
- Hennink WE, van Nostrum CF. Novel cross-linking methods to design hydrogels. Adv. Drug Deliv. Rev. 2002;54:13–36.10.1016/S0169-409X(01)00240-X
- Zhang Y, Zhu W, Ding J. Preparation of thermosensitive microgels via suspension polymerization using different temperature protocols. J. Biomed. Mater. Res. Part A. 2005;75A:342–349.10.1002/(ISSN)1552-4965
- Wang Zh, Hou X, Mao Zh, et al. Synthesis and characterization of biodegradable poly (lactic acid-co-glycine) via direct melt copolymerization. Iran. Polym. J. 2008;17:791–798.
- Bagheri Sh, Mohammadi-Rovshandeh J, Hassan A. Synthesis and characterization of biodegradable random copolymers of L-lactide, glycolide and trimethylene carbonate. Iran. Polym. J. 2007;16:489–494.
- Lopes MA, Felisberti MI. Mechanical behaviour and biocompatibility of poly(1-vinyl-2-pyrrolidinone)–gelatin IPN hydrogels. Biomaterials. 2003;24:1279–1284.10.1016/S0142-9612(02)00448-9
- Oh EJ, Kang SW, Kim BS, et al. Control of the molecular degradation of hyaluronic acid hydrogels for tissue augmentation. J. Biomed. Mater. Res. Part A. 2008;86A:685–693.10.1002/jbm.a.v86a:3
- Coviello T, Grassi M, Rambone G, et al. A crosslinked system from Scleroglucan derivative: preparation and characterization. Biomaterials. 2001;22:1899–1909.10.1016/S0142-9612(00)00374-4
- Garcia Y, Collighan R, Griffin M, et al. Assessment of cell viability in a three-dimensional enzymatically cross-linked collagen scaffold. J. Mater. Sci. Mater. Med. 2007;18:1991–2001.10.1007/s10856-007-3091-9
- Baroli B. Photopolymerization in drug delivery, tissue engineering and cell encapsulation: issues and potentialities. J. Chem. Tech. Biotech. 2006;81:491–499.10.1002/(ISSN)1097-4660
- Kimura M, Fukumoto K, Watanabe J, et al. Hydrogen-bonding-driven spontaneous gelation of water-soluble phospholipid polymers in aqueous medium. J. Biomater. Sci. Polym. Ed. 2004;15:631–644.10.1163/156856204323046898
- Oh KS, Han SK, Choi YW, et al. Hydrogen-bonded polymer gel and its application as a temperature-sensitive drug delivery system. Biomaterials. 2004;25:2393–2398.10.1016/j.biomaterials.2003.09.008
- Stenekes RJ, Talsma H, Hennink WE. Formation of dextran hydrogels by crystallization. Biomaterials. 2001;22:1891–1898.10.1016/S0142-9612(00)00375-6
- Cappello J, Crissman JW, Crissman M, et al. In-situ self-assembling protein polymer gel systems for administration, delivery, and release of drugs. J. Controlled Release. 1998;53:105–117.
- Ganji F, Abdekhodaie MJ, Ramazany-Sadtabadi A. Gelation time and degradation rate of chitosan as a thermosensitive injectable hydrogel. J. Sol-Gel. Sci. Technol. 2007;42:47–53.10.1007/s10971-006-9007-1
- Mohamadnia Z, Jamshidi A, Mobedi H, et al. Full natural hydrogel beads for controlled release of acetate and disodium phosphate derivatives of betamethasone. Iran. Polym. J. 2007;16:711–718.
- Aalaie J, Vasheghani-Farahani E, Rahmatpour A, et al. Effect of montmorillonite on gelation and swelling behavior of sulfonated polyacrylamide nanocomposite hydrogels in electrolyte solutions. Eur. Polymer J. 2008;44:2024–2031.10.1016/j.eurpolymj.2008.04.031
- Qiu Y, Park K. Environment-sensitive hydrogels for drug delivery. Adv. Drug Deliv. Rev. 2012;64:49–60.10.1016/j.addr.2012.09.024
- Baroli B. Hydrogels for tissue engineering and delivery of tissue-inducing substances. J. Pharm. Sci. 2007;96:2197–2223.10.1002/jps.20873
- Entezami AA, Massoumi B. Artificial muscles, biosensors and drug delivery systems based on conducting polymers: a review. Iran. Polym. J. 2006;15:13–30.
- Schmaljohann D. Thermo- and pH-responsive polymers in drug delivery. Adv. Drug Deliv. Rev. 2006;58:1655–1670.10.1016/j.addr.2006.09.020
- Casadei MA, Pitarresi G, Calabrese R, et al. Biodegradable and pH-sensitive hydrogels for potential colon-specific drug delivery: characterization and in vitro release studies. Biomacromolecules. 2008;9:43–49.10.1021/bm700716c
- Chiu HC, Wu AT, Lin YF. Synthesis and characterization of acrylic acid-containing dextran hydrogels. Polymer. 2001;42:1471–1479.10.1016/S0032-3861(00)00523-1
- Tavakoli J, Jabbari E, Etrati Khosroshahi M, et al. Swelling characterization of anionic acrylic acid hydrogel in an external electric field. Iran. Polym. J. 2006;15:891–900.
- Lim F, Sun AM. Microencapsulated islets as bioartificial pancreas. Science. 1980;210:908–910.10.1126/science.6776628
- Yannas, Lee E, Orgill DP. Synthesis and characterization of a model extracellular matrix that induces partial regeneration of adult mammalian skin. Proc. Natl. Acad. Sci. USA. 1989;86:933–937.
- Gin H, Dupuy B, Baquey A, et al. Lack of responsiveness to glucose of microencapsulated Islets of Langerhans after three weeks implantation in the rat – influence of complement. J. Microencapsul. 1990;7:341–346.10.3109/02652049009021844
- Matthew HW, Salley SO, Peterson WD, et al. Complex coacervate microcapsules for mammalian cell culture and artificial organ development. Biotechnol. Prog. 1993;9:510–519.10.1021/bp00023a010
- Hsu FY, Tsai SW, Wang FF, et al. The collagen-containing alginate/poly (L-lysine)/alginate microcapsules. Artif. Cells Blood Substit. Immobil. Biotechnol. 2000;28(2):147–154.
- Sefton MV, May MH, Lahooti S, et al. Making microencapsulation work: conformal coating immobilization gels and in vivo performance. J. Controlled Release. 2000;65:173–186.10.1016/S0168-3659(99)00234-5
- Woerly S. Porous hydrogels for neural tissue engineering. Porous. Mater. Tiss. Eng. 1997;250:53–68.
- Hubbell JA. Synthetic biodegradable polymers for tissue engineering and drug delivery. Curr. Opin. Solid State Mater. Sci. 1998;3:246–251.10.1016/S1359-0286(98)80098-3
- Dillon JP, Yu XJ, Sridharan A, et al. The influence of physical structure and charge on neurite extension in a 3D hydrogel scaffold. J. Biomater. Sci. Polym. Ed. 1998;9:1049–1069.10.1163/156856298X00325
- Cao YL, Rodriguez A, Vacanti M, et al. Comparative study of the use of PGA, calcium alginate and pluronics in the engineering of autologous porcine cartilage. J. Biomater. Sci. Polym. Ed. 1998;9:475–487.
- Borkenhagen M, Clemence JF, Sigrist H, et al. Three-dimensional extracellular matrix engineering in the nervous system. J. Biomed. Mater. Res. 1998;40:392–400.10.1002/(ISSN)1097-4636
- Kim BS, Nikolovski J, Bonadio J, et al. Cyclic mechanical strain regulates the development of engineered smooth muscle tissue. Nat. Biotechnol. 1999;17:979–983.10.1038/13671
- Elisseeff J, Anseth K, Sims D, et al. Transdermal photopolymerization for minimally invasive implantation. Proc. Nat. Acad. Sci. 1999;96:3104–3107.10.1073/pnas.96.6.3104
- Suggs LJ, Mikos AG. Development of poly (propylene fumarate-co-ethylene glycol) as an injectable carrier for endothelial cells. Cell. Transplant. 1999;8:345–350.
- Campoccia D, Doherty P, Radice M, et al. Semisynthetic resorbable materials from hyaluronan esterification. Biomaterials. 1998;19:2101–2127.
- Prestwich GD, Marecak DM, Marecak JF, et al. Controlled chemical modification of hyaluronic acid. J. Controlled Release. 1998;53:93–103.10.1016/S0168-3659(97)00242-3
- Nakamae K, Miyata T, Jikihara A, et al. Formation of poly (glucosyloxyethyl methacrylate)–concanavalin A complex and its glucose sensitivity. J. Biomater. Sci. Polym. Ed. 1994;6:79–90.
- Morris JE, Fischer R, Hoffman AS. Affinity precipitation of proteins with polyligands. Anal. Biochem. 1993;41:991–997.
- Drumheller P, Hubbell JA. Densely crosslinked polymer networks of PEG in trimethylolpropane triacrylate for cell adhesion-resistant surfaces. J. Biomed. Mater. Res. 1995;29:201–215.
- Fariba G, Ebrahim VF. Hydrogels in controlled drug delivery systems. Iran. Polym. J. 2009;18:63–88.
- Lowman AM, Peppas NA. Hydrogels. In: E Mathiowitz, editor. Encyclopedia of controlled drug delivery. New York, NY: Wiley; 1999. p. 397–418.
- Ratner BD, Hoffman AS. Synthetic hydrogels for biomedical applications. In: JD Andrade, editor. Hydrogels for medical and related applications, ACS symposium series, Vol. 31. Washington, DC: American Chemical Society; 1976. p. 1–36.10.1021/symposium
- Peppas NA, Mikos AG. Preparation methods and structure of hydrogels. In: NA Peppas, editor. Hydrogels in medicine and pharmacy, Vol. I. Boca Raton, FL: CRC Press; 1986. p. 1–26.
- Am Ende MT, Mikos AG. Diffusion-controlled delivery of proteins from hydrogels and other hydrophilic systems. In: LM Sanders, RW Hendren, editors. Protein delivery: physical systems. Tokyo: Plenum Press; 1997. p. 139–165.
- Peppas NA, Barr-Howell BD. Characterization of the cross-linked structure of hydrogels. In: NA Peppas, editor. Hydrogels in medicine and pharmacy, Vol. I. Boca Raton, FL: CRC Press; 1986. p. 27–56.
- Britton LN, Ashman RB, Aminahhavi TM, et al. Prediction of transport properties of permeants through polymer films: a simple gravimetric experiment. J. Chem. Educ. 1988;65:368.10.1021/ed065p368
- Peppas NA, Huang Y, Torres-Lugo M, et al. Physicochemical foundations and structural design of hydrogels in medicine and biology. Annu. Rev. Biomed. Eng. 2000;2:9–29.10.1146/annurev.bioeng.2.1.9
- Canal T, Peppas NA. Correlation between mesh size and equilibrium degree of swelling of polymeric network. J. Biomed. Mater. Res. 1989;23:1183–1193.10.1002/(ISSN)1097-4636
- Ichi T, Watanabe J, Ooya T, et al. Design of hydrogels crosslinked by biodegradable polyrotaxanes. Trans. Soc. Biomater. 2000; 33:1438.
- Clapper DL, Burkstrand MJ, Chudzik SJ, et al. A photo-crosslinked collagen/BMP matrix promotes bone formation in vivo. Trans. Soc. Biomater. 2000; 25:115–492.
- Thompson JP, Oegema TR, Jr, Bradford DS. Stimulation of mature canine intervertebral disc by growth factors. Spine (Phila Pa 1976). 1991; 16(3):253–260.
- Dong LC, Hoffman AS, Yan Q. Macromolecular penetration through hydrogels. J. Biomater. Sci. Polym. Ed. 1994;5:473–484.10.1163/156856294X00158
- Kuo CK, Ma PX. Diffusivity of three-dimensional ionically-crosslinked alginate hydrogels. Polym. Prep. 2000;41:1661.
- Marler JJ, Guha A, Rowley J, et al. Soft tissue augmentation with injectable alginate and syngeneic fibroblasts. Plast. Reconstr. Surg. 2000;105:2049–2058.10.1097/00006534-200005000-00020
- Ranjha NM, Khan S. Chitosan/poly (vinyl alcohol) based hydrogels for biomedical applications; A review. J. Pharm. Altern. Med. 2013;2:30–41.
- Hoare TR, Kohane DS. Hydrogels in drug delivery: progress and challenges. Polymer. 2008;49:1993–2007.10.1016/j.polymer.2008.01.027
- Berger J, Reist M, Mayer JM, et al. Structure and interactions in covalently and ionically crosslinked chitosan hydrogels for biomedical applications. Eur. J. Pharm. Biopharm. 2004;57:19–34.10.1016/S0939-6411(03)00161-9
- Hennink WE, van Nostrum CF. Novel crosslinking methods to design hydrogels. Adv. Drug Deliv. Rev. 2002;54:13–36.10.1016/S0169-409X(01)00240-X
- Kuijpers AJ, Engers GHM, Meyvis TKL, et al. Combined gelatin-chondroitin sulfate hydrogels for controlled release of cationic antibacterial proteins. Macromolecules. 2000; 33:3705–3713.
- Eiselt P, Lee KY, Mooney DJ. Rigidity of two-component hydrogels prepared from alginate and poly (ethylene glycol)-diamines. Macromolecules. 1999;32:5561–5566.10.1021/ma990514m
- de Nooy AEJ, Masci G, Crescenzi V. Versatile synthesis of polysaccharide hydrogels using the Passerini and Ugi multicomponent condensations. Macromolecules. 1999;32:1318–1320.10.1021/ma9815455
- de Nooy AEJ, Capitani D, Masci G, et al. Ionic polysaccharide hydrogels via the Passerini and Ugi multicomponent condensations: synthesis, behavior and solid-state NMR characterization. Biomacromolecules. 2000;1:259–267.10.1021/bm005517h
- Ichi T, Watanabe J, Ooya T, et al. Controllable erosion time and profile in poly(ethylene glycol) hydrogels by supramolecular structure of hydrolysable polyrotaxane. Biomacromolecules. 2001;2:204–210.10.1021/bm005617n
- Tan H, Chu CR, Payne KA, et al. Injectable in situ forming biodegradable chitosan-hyaluronic acid based hydrogels for cartilage tissue engineering. Biomaterials. 2009;30:2499–2506.10.1016/j.biomaterials.2008.12.080
- Lee KY, Alsberg E, Mooney DJ. Degradable and injectable poly (aldehyde guluronate) hydrogels for bone tissue engineering. J. Biomed. Mater. Res. 2001;56:228–233.10.1002/(ISSN)1097-4636
- Elbert DL, Pratt AB, Lutolf MP, et al. Protein delivery from materials formed by self-selective conjugate addition reactions. J. Controlled Release. 2001;76:11–25.10.1016/S0168-3659(01)00398-4
- Giammona G, Pitarresi G, Cavallaro G, et al. New biodegradable hydrogels based on an acryloylated polyaspartamide crosslinked by gamma irradiation. J. Biomater. Sci. Polym. Ed. 1999;10:969–987.10.1163/156856299X00568
- Hiemstra C, van der Aa LJ, Zhong ZY, et al. Novel in situ forming, degradable dextran hydrogels by Michael addition chemistry: synthesis, rheology, and degradation. Macromolecules. 2007;40:1165–1173.10.1021/ma062468d
- Giammona G, Pitarresi G, Cavallaro G, et al. New biodegradable hydrogels based on an acryloylated polyaspartamide cross-linked by gamma irradiation. J. Biomater. Sci. Polym. Ed. 1999;10:969–987.10.1163/156856299X00568
- Martens P, Anseth KS. Characterization of hydrogels from acrylate modified poly (vinyl alcohol) macromers. Polymer. 2000;41:7715–7722.10.1016/S0032-3861(00)00123-3
- Peppas NA, Mikos AG. Preparation methods and structure of hydrogels. In: NA Peppas, editor. Hydrogels in medicine and pharmacy, Vol. I. Boca Raton, FL: CRC Press; 1986. Chapter 1; p. 1–27.
- Peppas NA, Merrill EW. Hydrogels as swollen elastic networks. J. Appl. Polym. Sci. 1977;21:1763–1770.10.1002/app.1977.070210704
- Kofinas P, Athanasssiou V, Merrill EW. Hydrogels prepared by electron irradiation of poly (ethylene oxide) in water solution: unexpected dependence of density and protein diffusion coefficients on initial PEO molecular weight. Biomaterials. 1996;17:1547–1550.10.1016/0142-9612(96)89781-X
- Merrill EW, Dennison KA, Sung C. Partitioning and diffusion of solutes in hydrogels of poly (ethylene oxide). Biomaterials. 1993;14:1117–1126.10.1016/0142-9612(93)90154-T
- Stringer JL, Peppas NA. Diffusion of small molecular weight drugs in radiation-crosslinked poly (ethylene oxide) hydrogels. J. Controlled Release. 1996;42:195–202.10.1016/0168-3659(96)01457-5
- Jabbari E, Nozari S. Swelling behavior of acrylic acid hydrogels prepared by γ-radiation crosslinking of polyacrylic acid in aqueous solution. Eur. Polymer J. 2000;36:2685–2692.10.1016/S0014-3057(00)00044-6
- Moerkerke R, Meeussen F, Koningsveld R, et al. Phase transitions in swollen networks. 3. Swelling behavior of radiation crosslinked poly (vinyl methyl ether) in water. Macromolecules. 1998;31:2223–2229.
- Arndt KF, Schmidt T, Reichelt R. Thermo-sensitive poly (vinyl methyl ether) micro-gel formed by high energy radiation. Polymer. 2001;42:6785–6791.10.1016/S0032-3861(01)00164-1
- Sperinde JJ, Griffith LG. Synthesis and characterization of enzymatically-crosslinked-poly (ethylene glycol) hydrogels. Macromolecules. 1997;30:5255–5264.10.1021/ma970345a
- Sperinde JJ, Griffith LG. Control and predication of gelation kinetics in enzymatically cross-linked poly-(ethylene glycol) hydrogels. Macromolecules. 2000;33:5467–5480.
- Lim DW, Nettles DL, Setton LA, et al. Rapid cross-linking of elastin-like polypeptides with (hydroxymethyl)phosphines in aqueous solution. Biomacromolecules. 2007;8:1463–1470.10.1021/bm061059m
- Wu J, Su ZG, Ma GH. A thermo- and pH-sensitive hydrogel composed of quaternized chitosan/glycerophosphate. Int. J. Pharm. 2006;315:1–11.10.1016/j.ijpharm.2006.01.045
- Yokoyama F, Masada I, Shimamura K, et al. Morphology and structure of highly elastic poly (vinyl alcohol) hydrogel prepared by repeated freezing-and- melting. Colloid. Polym. Sci.. 1986;264:595–601.10.1007/BF01412597
- Tsuji H. Poly(lactide) stereocomplexes: formation, structure, properties, degradation, and applications. Macromol. Biosci. 2005;5:569–597.10.1002/(ISSN)1616-5195
- Ikada Y, Jamshidi K, Tsuji H, et al. Stereocomplex formation between enantiomeric poly (lactides). Macromolecules. 1987;20:904–906.10.1021/ma00170a034
- Alexander C. Temperature-and pH-responsive smart polymers for gene delivery. Expert Opin. Drug Deliv. 2006;3:573–581.10.1517/17425247.3.5.573
- Bignotti F, Penco M, Sartore L, et al. Synthesis, characterisation and solution behavior of thermo-and pH-responsive polymers bearing l-leucine residues in the side chains. Polymer. 2000; 41: 8247-8256.
- Garbern JC, Hoffman AS, Stayton PS. Injectable pH-and temperature-responsive poly (N-isopropylacylamide-co-propylacrylic acid) copolymers for delivery of angiogenic growth factors. Biomacromolecules. 2010;11:1833–1839.10.1021/bm100318z
- Jeong B, Gutowska A. Lessons from nature: stimuli-responsive polymers and their biomedical applications. Trends Biotechnol. 2002;20:305–311.10.1016/S0167-7799(02)01962-5
- Tomme V, Storm SRG, Hennink WE. In situ gelling hydrogels for pharmaceutical and biomedical applications. Int. J. Pharm. 2008;355:1–18.10.1016/j.ijpharm.2008.01.057
- Fogueri LR, Singh S. Smart polymers for controlled delivery of proteins and peptides: a review of patents. Recent. Pat. Drug Deliv. Formul. 2009; 3:40–48.
- Ruel-Gariepy E, Leroux JC. In situ-forming hydrogels – review of temperature-sensitive systems. Eur. J. Pharm. Biopharm. 2004;58:409–426.10.1016/j.ejpb.2004.03.019
- Liu F, Urban MW. Recent advances and challenges in designing stimuli-responsive polymers. Prog. Polym. Sci. 2010;35:3–23.10.1016/j.progpolymsci.2009.10.002
- Gil ES, Hudson SM. Stimuli-responsive polymers and their bioconjugates. Prog. Polym. Sci. 2004;29:1173–1222.10.1016/j.progpolymsci.2004.08.003
- Bajpai A, Shukla SK, Bhanu S, et al. Responsive polymers in controlled drug delivery. Prog. Polym. Sci. 2008;33:1088–1118.10.1016/j.progpolymsci.2008.07.005
- Dai S, Ravi P, Tam KC. pH-Responsive polymers: synthesis, properties and applications. Soft Matter. 2008;4:435–449.10.1039/b714741d
- Ravaine V, Ancla C, Catargi B. Chemically controlled closed-loop insulin delivery. J. Controlled Release. 2008;132:2–11.10.1016/j.jconrel.2008.08.009
- Roy VD, Cambre JN, Sumerlin BS. Future perspectives and recent advances in stimuli-responsive materials. Prog. Polym. Sci. 2010;35:278–301.10.1016/j.progpolymsci.2009.10.008
- Tanaka IN, Nishio I, Sun ST, et al. Collapse of gels in an electric field. Science. 1982;218:467–469.10.1126/science.218.4571.467
- JP, Nitta T, Osada Y. Electro kinetic modeling of the contractile phenomena of polyelectrolyte gels. One-dimensional capillary model. J. Phys. Chem. 1994; 98: 9583–9587.
- Sawahata K, Hara M, Yasunaga H, et al. Electrically controlled drug delivery system using polyelectrolyte gels. J. Controlled Release. 1990;14:253–262.10.1016/0168-3659(90)90165-P
- Kwon IC, Bae YH, Okano T, et al. Drug release from electric current sensitive polymers. J. Controlled Release. 1991;17:149–156.10.1016/0168-3659(91)90054-H
- Mamada A, Tanaka T, Kungwachakun D, et al. Photo induced phase transition of gels. Macromolecules. 1990;23:1517–1519.10.1021/ma00207a046
- Suzuki A, Tanaka T. Phase transition in polymer gels induced by visible light. Nature. 1990;346:345–347.10.1038/346345a0
- Lee KK, Cussler EL, Marchetti M, et al. Pressure-dependent phase transitions in hydrogels. Chem. Eng. Sci. 1990;45:766–767.10.1016/0009-2509(90)87019-O
- Zhong X, Wang YX, Wang SC. Pressure dependence of the volume phase transition of temperature-sensitive gels. Chem. Eng. Sci. 1996;51:3235–3239.10.1016/0009-2509(95)00344-4
- Miyata T, Asami N, Uragami T. A reversibly antigen-responsive hydrogel. Nature. 1999;399:766–769.
- Suzuki Y, Tanihara M, Nishimura Y, et al. A new drug delivery system with controlled release of antibiotic only in the presence of infection. J. Biomed. Mater. Res. 1998;42:112–116.10.1002/(ISSN)1097-4636
- Tanihara M, Suzuki Y, Nishimura Y, et al. Thrombin-sensitive peptide linkers for biological signal-responsive drug release systems. Peptides. 1998;19:421–425.10.1016/S0196-9781(97)00420-8
- Tanihara M, Suzuki Y, Nishimura Y, et al. A novel microbial infection-responsive drug release system. J. Pharm. Sci. 1999;88:510–514.10.1021/js980418j
- Mohamadnia Z, Zohuriaan-Mehr AJ, Kabiri K, et al. Bioactive compatible. Polymer. 2007;22:342–356.
- Yin LC, Fei LK, Cui FY, et al. Superporous hydrogels containing poly(acrylic acid-co-acrylamide)/O-carboxymethyl chitosan interpenetrating polymer networks. Biomaterials. 2007;28:1258–1266.10.1016/j.biomaterials.2006.11.008
- Zhang YX, Wu FP, Li MZ, et al. pH switching on-off semi-IPN hydrogel based on cross-linked poly(acrylamide-co-acrylic acid) and linear polyallyamine. Polymer. 2005;46:7695–7700.10.1016/j.polymer.2005.05.121
- Ankareddi I, Brazel CS. Int. J. Pharm. 2007;336:241–247.10.1016/j.ijpharm.2006.11.065
- Matsusaki M, Sakaguchi H, Serizawa T, et al. Controlled release of vascular endothelial growth factor from alginate hydrogels nano-coated with polyelectrolyte multilayer films. J. Biomater. Sci. Polym. Ed. 2007;18:775–783.10.1163/156856207781034160
- De Geest BG, Dejugnat C, Sukhorukov GB, et al. Self-rupturing microcapsules. Adv. Mater. 2005;17:2357–2361.10.1002/(ISSN)1521-4095
- Buenger D, Topuz F, Groll J. Hydrogels in sensing applications. Prog. Polym. Sci. 2012;37:1678–1719.10.1016/j.progpolymsci.2012.09.001
- Nguyen KT, West JL. Photopolymerizable hydrogels for tissue engineering applications. Biomaterials. 2002;23:4307–4314.10.1016/S0142-9612(02)00175-8
- Biswal D, Hilt JZ. Microscale analysis of patterning reactions via FTIR imaging: application to intelligent hydrogel systems. Polymer. 2006;47:7355–7360.10.1016/j.polymer.2006.08.028
- Dalton PD, Hostert C, Albrecht K, et al. Structure and properties of urea-crosslinked star poly (ethylene oxide)-ran-(propylene oxide) hydrogels. Macromol. Biosci. 2008;8:923–931.10.1002/mabi.v8:10
- Kopecek J, Yang JY. Peptide-directed self-assembly of hydrogels. Acta. Biomater. 2009;5:805–816.
- Vandermeulen GWM, Tziatzios C, Duncan R, et al. PEG-based hybrid block copolymers containing alpha-helical coiled coil peptide sequences: control of self-assembly and preliminary biological evaluation. Macromolecules. 2005;38:761–769.10.1021/ma0485538
- Toledano S, Williams RJ, Jayawarna V, et al. Enzyme-triggered self-assembly of peptide hydrogels via reversed hydrolysis. J. Am. Chem. Soc. 2006;128:1070–1071.10.1021/ja056549l
- Galler KM, Aulisa L, Regan KR, et al. Self-assembling multidomain peptide hydrogels: designed susceptibility to enzymatic cleavage allows enhanced cell migration and spreading. J. Am. Chem. Soc. 2010;132:3217–3223.10.1021/ja910481t
- Lin CC, Metters AT, Anseth KS. Functional PEG-peptide hydrogels to modulate local inflammation induced by the pro-inflammatory cytokine TNF alpha. Biomaterials. 2009;30:4907–4914.10.1016/j.biomaterials.2009.05.083
- Dankers PYW, Harmsen MC, Brouwer LA, et al. A modular and supramolecular approach to bioactive scaffolds for tissue engineering. Nat. Mater. 2005;4:568–574.10.1038/nmat1418
- Um SH, Lee JB, Park N, et al. Enzyme-catalysed assembly of DNA hydrogel. Nat. Mater. 2006;5:797–801.10.1038/nmat1741
- Horkay F, Basser PJ. Osmotic observations on chemically crosslinked DNA gels in physiological salt solutions. Biomacromolecules. 2004;5:232–237.10.1021/bm034372m
- Yoshida R, Uchida K, Kaneko Y, et al. Comb-type grafted hydrogels with rapid de-swelling response to temperature changes. Nature. 1995;374:240–242.10.1038/374240a0
- Irie M, Misumi Y, Tanaka T. Stimuli-responsive polymers – chemical induced reversible separation of an aqueous solution of poly (N-isopropylacrylamide) with pendent crown ether groups. Polymer. 1993;34:4531–4535.10.1016/0032-3861(93)90160-C
- Lee KK, Cussler EL, Marchetti M, et al. Pressure-dependent phase-transitions in hydrogels. Chem. Eng. Sci. 1990;45:766–767.10.1016/0009-2509(90)87019-O
- Bohon K, Krause S. An electrorheological fluid and siloxane gel based electromechanical actuator: working toward an artificial muscle. J. Polym. Sci. Part B: Polym. Phys. 1998; 36:1091–1094.
- Horkay F, Tasaki I, Basser PJ. Osmotic swelling of polyacrylate hydrogels in physiological salt solutions. Biomacromolecules. 2000;1:84–90.10.1021/bm9905031
- Gupta KM, Barnes SR, Tangaro RA, et al. Temperature and pH sensitive hydrogels: an approach towards smart semen-triggered vaginal microbicidal vehicles. J. Pharm. Sci. 2007;96:670–681.10.1002/jps.20752
- Zhao B, Moore JS. Fast pH- and ionic strength-responsive hydrogels in microchannels. Langmuir. 2001;17:4758–4763.10.1021/la001709m
- Ju XJ, Chu LY, Liu L, et al. A novel thermoresponsive hydrogel with ion-recognition property through supramolecular host-guest complexation. J. Phys. Chem. B. 2008;112:1112–1118.10.1021/jp709746w
- Hassan CM, Doyle FJ, Peppas NA. Dynamic behavior of glucose responsive poly (methacrylic acid-g-ethylene glycol) hydrogels. Macromolecules. 1997;30:6166–6173.10.1021/ma970117g
- Miyata T, Asami N, Uragami T. A reversibly antigen-responsive hydrogel. Nature. 1999;399:766–769.
- Tessmar JK, Gopferich AM. Matrices and scaffolds for protein delivery in tissue engineering. Adv. Drug Deliv. Rev. 2007;59:274–291.10.1016/j.addr.2007.03.020
- Holland TA, Tessmar JK, Tabata Y, et al. Transforming growth factor-beta 1 release from oligo poly (ethylene glycol) fumarate) hydrogels in conditions that model the cartilage wound healing environment. J. Controlled Release. 2004;94:101–114.10.1016/j.jconrel.2003.09.007
- Kohane DS, Langer R. Polymeric biomaterials in tissue engineering. Pediatr. Res. 2008;63:487–491.10.1203/01.pdr.0000305937.26105.e7
- Kim SW, Bae YH, Okano T. Hydrogels: swelling, drug loading, and release. Pharm. Res. 1992;9:283–290.10.1023/A:1015887213431
- Lin CC, Metters AT. Hydrogels in controlled release formulations: network design and mathematical modeling. Adv. Drug Deliv. Rev. 2006;58:1379–1408.10.1016/j.addr.2006.09.004
- Hoare TR, Kohane DS. Hydrogels in drug delivery: progress and challenges. Polymer. 2008;49:1993–2007.10.1016/j.polymer.2008.01.027
- Petersen LK, Narasimhan B. Combinatorial design of biomaterials for drug delivery: opportunities and challenges. Expert Opin. Drug Deliv. 2008;5:837–846.10.1517/17425247.5.8.837
- Lutolf MP, Hubbell JA. Synthetic biomaterials as instructive extracellular microenvironments for morphogenesis in tissue engineering. Nat. Biotechnol. 2005;23:47–55.10.1038/nbt1055
- Lee KY, Mooney DJ. Hydrogels for tissue engineering. Chem. Revs. 2001;101:1869–1879.
- Hutmacher DW. Scaffold design and fabrication technologies for engineering tissues – state of the art and future perspectives. J. Biomater. Sci. Polym. Ed. 2001;12:107–124.10.1163/156856201744489