
ABSTRACT
Hemoglobin Santa Ana [β88(F4)Leu→Pro (CTG > CCG) HBB: c.266T > C] is an unstable hemoglobin variant characterized by a substitution of the amino acid leucine by proline at the 88th position of the β-globin chain. We for the first time identified this hemoglobin variant in a Chinese patient by capillary electrophoresis (CE). The proband was an 8-year-old boy with chronic anemia, brown urine and splenomegaly. He had been affected by moderate anemia, twice approaching a severe degree, that was attributed to infection. The CE result revealed an abnormal hemoglobin peak at electrophoretic zone 4 that correspond to the hemoglobin Santa Ana peak, and a CTG > CCG mutation at codon 88 of the β-globin gene was confirmed by DNA sequencing. To avoid misdiagnosis and genetic risks, a literature review of other unstable hemoglobins that migrate similarly to the hemoglobin Santa Ana was performed. Our findings indicate that hemoglobin Santa Ana can be clearly separated by CE, with accurate diagnosis depending on molecular analysis. This information will be useful for providing appropriate genetic counselling and for prenatal diagnosis.
Introduction
Hemoglobin variants comprise a group of hereditary hemoglobin diseases caused by mutations in globin genes that lead to structural changes in the hemoglobin molecule. Hemoglobin variants with various clinical manifestations. Unstable hemoglobins are important causes of hemolytic anemia, which is rare and often undiagnosed. Hemoglobin Santa Ana, an unstable hemoglobin variant, was first reported in 1968 [Citation1] and was later confirmed to be due to the HBB gene mutation c.266T > C [Citation2]. In hemoglobin Santa Ana, the amino acid leucine at the 88th position of the normal β-globin chain is replaced by proline, which disturbs not only the heme contact but also the integrity of the F helix [Citation3]. Several patients with hemoglobin Santa Ana have been described, but detection by capillary electrophoresis (CE) has never been reported [Citation1–6]. The clinical manifestations of cases are very similar, including hemolytic anemia, jaundice, brown urine and splenomegaly.
This is the first report of a Chinese case of hemoglobin Santa Ana. Hematological parameters and molecular analysis results were compared with those of previously reported cases. In addition, a literature review of other unstable hemoglobins that migrate in a manner similar to that of hemoglobin Santa Ana on the CE electropherogram was conducted.
Case report
The proband originated from Luochuan City, Shanxi Province, China; the parents were healthy and free from anemia. The proband was an 8-year-old boy who was referred to our hospital because he twice had moderate-to-severe anemia. The boy was born at 39+ weeks of pregnancy with a birth weight of 3200 g. At 7 days after birth, he was hospitalized with neonatal jaundice and a hemoglobin level of 194 g/L; he was re-hospitalized at 27 days after birth with neonatal pneumonia and a hemoglobin level of 162 g/L. Brown urine was noted within several months after birth. Unexplained hemolytic anemia was found at 1 year old; his hemoglobin level was 90∼110 g/L. No specific treatment was performed at that time. Pulmonary infection occurred at the age of 2 years, which aggravated his anemia, and his hemoglobin level decreased to 69 g/L. Moreover, the boy manifested jaundice and soy sauce-coloured urine, and his spleen was slightly enlarged to 20 mm below the ribs. After recovering from the pulmonary infection, his hemoglobin level gradually restored and remained in the 90–100 g/L range. Unfortunately, the boy developed Epstein–Barr virus infection at the age of 7 years, and the degree of anemia worsened to moderate anemia, with a hemoglobin level progressively decreasing to 66 g/L; he then received a transfusion treatment of packed red blood cells. His spleen was significantly enlarged, and CT examination during hospitalization revealed multiple low-density areas in the organ. Bone marrow puncture indicated proliferative anemia. Some clinical blood and biochemical tests were performed during previous medical treatments in other hospitals. The causes of his anemia and splenomegaly were unknown. They agreed to participate in our study and signed informed consent forms. All studies were approved by the Ethics Committee of Guangdong Women and Children Hospital. To investigate aetiology, we carried out hematological parameter analysis, hemoglobin analysis and molecular diagnosis for family members.
Materials and methods
Hematological parameters were determined using a Sysmex XN5000 automated haematology analyser (Sysmex Corporation, Kobe, Japan). Hemoglobin analysis was conducted with an automated capillary electrophoresis system (Capillary 2, Sebia, France). Genomic DNA was extracted from peripheral blood leukocytes using the Lab-Aid 820 automation system (Zee San Biotech Company, China). Twenty-three common α- and β-thalassemia mutations in southern Chinese individuals were routinely measured in our laboratory by a suspension-array system, as previously described [Citation7]. To detect hemoglobin variants, PCR amplification of the α- and β-globin genes was carried out, and direct DNA sequencing of the amplified products was performed using an ABI PRISM 3130XL Analyser (Applied Biosystems, USA). The Power Plex® 21 System (Promega, USA) was employed to confirm genetic relationships. PCR products were analysed using ABI 3500xL Dx Genetic Analyzer (Applied Biosystems, USA).
Results
The hematological parameters and genotypes of the family members are listed in . Hematological parameters showed that the proband was anemiac, with a hemoglobin level of 94 g/L. A decreased mean corpuscular hemoglobin concentration (MCHC) and increased reticulocyte count were useful evidence of hemolytic anemia. According to CE, an abnormal hemoglobin X peak with a value of 6.6% of total hemoglobin was observed at electrophoretic zone 4, located between the hemoglobin F (Hb F) peak and hemoglobin A2 (Hb A2) peak (). No common α- and β-thalassemia mutations were found. Sanger sequencing for the proband revealed heterozygosity for a CTG > CCG mutation at codon 88 of the β-globin gene (HBB: c.266T > C) (), which corresponds to hemoglobin Santa Ana. No other mutations of clinical significance were observed in the α1- and α2-globin genes. All results for the proband's parents were normal and negative for hemoglobinopathies. The genetic relationship between the proband and his parents was verified. Therefore, this variant found in the proband was inferred to be a de novo mutation.
Figure 1. The CE result for the proband shows the Hb X fraction, which corresponds to the hemoglobin Santa Ana peak.
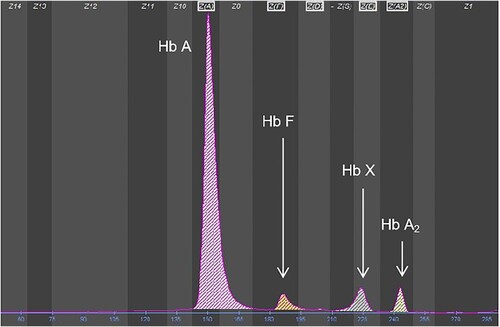
Figure 2. The result of Sanger sequencing. The arrow indicates the CTG > CCG mutation at codon 88 of the HBB gene previously reported as hemoglobin Santa Ana.
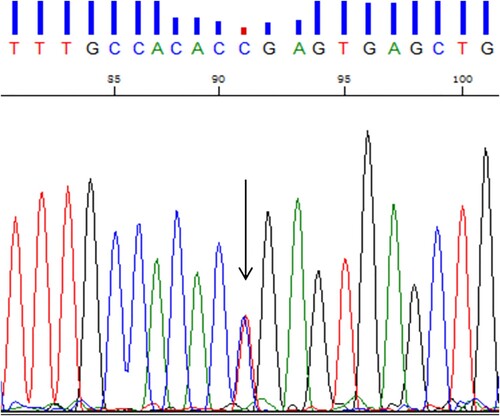
Table 1. Hematological and genotypic data of the family members.
The hemoglobin Santa Ana peak at zone 4 migrated similarly to hemoglobin E (Hb E) (). In addition, 5 other unstable hemoglobins migrated in the same zone as hemoglobin Santa Ana, all of which are β-chain variants. A list of these unstable hemoglobins is provided in . Based on previous studies [Citation8,Citation9], the degree of relative instability, change in oxygen affinity, clinical presentation and CE finding of each heterozygous variant were reviewed.
Table 2. Unstable hemoglobin variants migrated at zone 4 on the CE electrophoregram.
Discussion and literature review
Unstable hemoglobins are a class of structurally abnormal hemoglobin variants. Various mutations in one of the globin-coding genes can produce unstable hemoglobin, which can alter the structure of the hemoglobin tetramer, leading to its instability, altered solubility, and intracellular precipitation [Citation9–11]. More than 150 unstable hemoglobins are listed in the comprehensive hemoglobin variant database (http://globin.cse.psu.edu./hbvar/menu.html) [Citation12,Citation13]. Hemoglobin Santa Ana is a rare unstable hemoglobin characterized by a substitution of leucine by proline at the 88th position of the normal β-globin chain. Although the proline residue in hemoglobin Santa Ana occurs in a helical portion of the molecule (F4), it may not necessarily cause a change in the conformation of the main chain. Of greater importance may be that this mutation disrupts the interaction between the hydrophobic side chain of leucine and protoheme, a change that permits water to enter the heme pocket, causing the formation of methaemoglobin and denaturation of globin and resulting in intracellular precipitation. Hemoglobin Santa Ana was first observed in 1968 by Opfell et al. in the United States, and several cases have since been described in Indian, Brazilian and Japanese families [Citation1–6]. Overall, the cases have similar clinical symptoms, including hemolytic anemia, jaundice, brown urine and splenomegaly. A hemolysis crisis may occur in the presence of infection or exposure to oxidant medications. However, blood transfusions are rarely required for these cases, and anemia can be improved after splenectomy. A similar clinical course was found in our patient, and splenectomy is being planned.
Most unstable hemoglobin variants follow an autosomal dominant pattern of inheritance, and spontaneous mutations have seldom been described[R10]. The hemoglobin Santa Ana variant arose as a de novo mutation in our case, as well as in some previously reported patients whose parents had normal hemoglobins [Citation1–3].
Both the detection and identification of unstable hemoglobins are clinically important. A normal hemoglobin result detected by hemoglobin electrophoresis or high-performance liquid chromatography (HPLC) does not rule out unstable hemoglobins because unstable hemoglobin undergoes rapid denaturation and degradation within the cell, with only normal hemoglobin being detected by testing. Occasionally, an unremarkable abnormal result may appear due to partial denaturation of hemoglobin [Citation10,Citation11]. According to previous reports, the ratio of hemoglobin Santa Ana to normal hemoglobin obtained by HPLC and electrospray ionization mass spectrometry were approximately 15% and 40%, respectively; this discrepancy is attributed to a loss of a heme portion per molecule, with the former calculated on the basis of heme and the latter on the basis of the β-subunit [Citation3,Citation5]. Our research confirms for the first time that hemoglobin Santa Ana can be detected and clearly separated by the CE method, even though the ratio (6.6%) was lower than the results obtained by previous methods (). CE also revealed an increased level of HbA2 (3.9%), which is a common finding for unstable β-chain hemoglobin.
Currently, discovery of abnormal hemoglobin relies on hemoglobin electrophoresis commonly used in clinical laboratories. Different hemoglobin molecules can be detected in the same electrophoresis zone position, akin to the hemoglobin Santa Ana peak migrating similar to Hb E in the present study, which may cause misjudgment or quantitative errors of electrophoresis bands. Therefore, some pathogenic abnormal hemoglobin can easily be missed or misdiagnosed. To obtain a better understanding of this issue, the other 5 types of unstable hemoglobins that migrate in the same zone as the hemoglobin Santa Ana were selected and reviewed. The severity of clinical presentation is quite variable in patients with unstable hemoglobin, ranging from asymptomatic to compensated hemolysis to severe anemia. Previous studies have also shown that clinical severity is linked to the influence of causative variants on hemoglobin stability and oxygen affinity. Unstable hemoglobins with high oxygen affinity tend to cause milder anemia because of compensatory erythropoiesis stimulated by erythropoietin [Citation10]. Unlike previous experiences, hemoglobin Santa Ana in a heterozygous state exhibits an extremely high affinity for oxygen but lead to a condition that appear close to moderate anemia. Our summary suggests that the degree of hemoglobin instability may be a significant determinant: the more unstable the hemoglobin structure is, the more severe are the symptoms that manifest. As shown in , for those with mildly unstable hemoglobin in the heterozygous state without anemia, remarkable abnormal fractions can appear by standard electrophoretic separation. Nevertheless, there are only subtle indications of abnormal fractions for severely unstable hemoglobins, regardless of oxygen affinity. Once an unstable β-globin variant accompanies β-thalassemia, the proportion of unstable hemoglobin will increase, with stronger adverse effects [Citation9]. In a previous report [Citation14], β-thalassemia accompanied by Hb Köln, an unstable β-globin hemoglobin, surprisingly resulted in apparently tolerated anemia. Given the above consideration, DNA sequencing should be performed for reliable identification of unstable hemoglobins.
Intrinsic disorders of erythrocytes are a common cause of hemolytic anemia in children and adults [Citation10]. Patients typically present symptoms of hemolytic anemia and dark urine should be needed to investigate this aspect. Due to the simple procedure, red cell enzyme assays should be performed preferentially. Once disorders of erythrocyte metabolism have been ruled out, unstable hemoglobinopathies should be considered. It is well known that the CE method is suitable for the detection and quantification of hemoglobin variants and thalassemias, with better performance than HPLC. In our study, we for the first time showed that hemoglobin Santa Ana can be detected by CE. This unstable hemoglobin is first described herein as a de novo variant in the Chinese population. In summary, our findings will be useful to assist in genetic counselling and prenatal diagnosis.
Declaration of conflicting interests
The authors declare that there is no conflict of interest.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Opfell RW, Lorkin PA, Lehmann H. Hereditary non-spherocytic haemolytic anaemia with post-splenectomy inclusion bodies and pigmenturia caused by an unstable haemoglobin Santa Ana-beta-88 (F4) leucine–proline. J Med Genet 1968;5(4):292–297.
- Gonçalves MS, Sonati MF, Kimura M, et al. Association of Hb santa ANA [α2β288(F4)LEU→PRO] And Hb porto alegre [α2β29(A6)SER→CYS] in a Brazilian female. Hemoglobin. 1994;18(3):235–239.
- Tanaka Y, Kelleher Jr JF, Schwartz E, et al. Oxygen binding and stability properties of Hb Santa Ana (β88 Leu→Pro). Hemoglobin. 1985;9(2):157–169.
- Fairbanks VF, Opfell RW, Burgert Jr EO. Three families with unstable hemoglobinopathies (köln. olmsted and Santa Ana) causing hemolytic anemia with inclusion bodies and pigmenturia. Am J Med. 1969;46(3):344–359.
- Miyazaki A, Nakanishi T, Kishikawa M, et al. The first Japanese case of Hb Santa Ana, an unstable abnormal hemoglobin, identified rapidly by electrospray ionization mass spectrometry. Intern Med. 1997;36(5):365–370.
- Robertson A, Rahemtulla A. Pulse oximetry error in a patient with a Santa Ana haemoglobinopathy. BMJ Case Rep. 2016;Sep 6;bcr2016216787: 392–399.
- Luo MY, Hu TT, Wang JC, et al. Application of a suspension array based on barcoded magnetic beads in thalassemia gene diagnosis. J Trop Med. 2016;16(7):832–835.
- Riou J, Szuberski J, Godart C, et al. Precision of CAPILLARYS 2 for the detection of hemoglobin variants based on their migration positions. Am J Clin Pathol 2018;149(2):172–180.
- Williamson D. The unstable haemoglobins. Blood Rev 1993;7(3):146–163.
- Gallagher PG. Diagnosis and management of rare congenital nonimmune hemolytic disease. Hematology. 2015; 2015: 392–399.
- Risinger M, Emberesh M, Kalfa TA, et al. Rare hereditary hemolytic anemias. Hematol Oncol Clin North Am 2019;33(3):373–392.
- Giardine B, Borg J, Viennas E, et al. Updates of the HbVar database of human hemoglobin variants and thalassemia mutations. Nucleic Acids Res. 2014;42(Database issue):D1063–D1069.
- Giardine BM, Joly P, Pissard S, et al. Clinically relevant updates of the HbVar database of human hemoglobin variants and thalassemia mutations. Nucleic Acids Res 2021;49(D1):D1192–D1196.
- Galacteros F, Loukopoulos D, Fessas P, et al. Hemoglobin koln occurring in association with a beta zero thalassemia: hematologic and functional consequences. Blood. 1989;74(1):496–500.