
ABSTRACT
Acute lymphoblastic leukemia (ALL) is a challenging disease with a growing genetic landscape, even though there is substantial gap between developed and non-developed countries when it comes to availability of such new technologies. This manuscript reports a 5-year retrospective cohort of newly diagnosed ALL patients and their genetic findings and outcomes. An expanded genetic evaluation by using FISH and RT–PCR was implemented, aiming to identify Ph-like alterations. Patients were treated according to our local protocol, which allocated patients according to age and Philadelphia-chromosome status. A total of 104 patients was included, with median age of 37.5 years. Philadelphia chromosome was detected in 33 cases of B-lineage. Among 45 Ph-negative B-lineage, after excluding KMT2A or TCF3-PBX1 cases, we identified 9 cases with Ph-like fusion. Ph-positive and Ph-like patients had higher initial WBC (p = 0.06). Out of 104 cases, two cases did not start chemotherapy and an early death rate of 10.8% was found. Allogeneic transplantation was performed in 18 cases, being ten performed in first CR. Three-year overall survival (OS) and 3-year event-free survival were 42.8% and 30.8%, respectively. For patients treated with a pediatric regimen, 3-year OS was 52.5%. Extramedullary disease (HR 0.42) and platelet counts (HR 0.9) were independently associated with OS. We still face excessive non-relapse mortality that compromises our results. Alternative strategies implementing FISH and RT–PCR are feasible and able to identify Ph-like fusions. Delays in allogeneic transplantation, as well as the unavailability of new agents, impact long-term survival. Measures to decrease early infection are desirable.
Introduction
Acute lymphoblastic leukemia (ALL) is a rare and challenging disease to manage, especially in adults, in which a higher number of high-risk genetic alterations are described and there is an increased treatment-related toxicity [Citation1]. Recent evidence has indicated overall survival (OS) rates around 50% in adults, with significant variability across risk stratification and consolidation strategies [Citation2,Citation3]. Over the last few years, newer genetic subtypes have been described, emphasizing the Philadelphia-like (Ph-like) signature, which is reported in 20-24% of adults with B-cell ALL [Citation4–6]. Among this subset of cases, fusions encompassing the CRLF2 gene comprise around 50% of cases [Citation7].
Continuous optimization of treatment regimens, adjusted to the local availability of infusion beds, newer drugs, and allogeneic hematopoietic transplantation (alloHCT), is warranted [Citation8]. In Latin America, patients newly diagnosed with ALL fare worse, mainly due to higher treatment-related toxicity as well as a supposedly greater incidence of Philadelphia-like cases in Hispanics [Citation9–12].
In our setting, a recent effort has been made to implement experience-based protocols and expand the genetic baseline assessment, ultimately aiming to improve cure rates. However, prior experience with a highly intensive protocol resulted in unacceptable toxicity and lower survival rates, leading our group to reconsider our strategy [Citation12]. While next-generation sequencing (NGS) or microarray platforms are not available in clinical routine for Ph-like screening, we started to evaluate suitable patients by using fluorescence in situ hybridization (FISH) and real-time polymerase chain reaction (RT–PCR) techniques, helping to segregate patients by their intrinsic genetic risk. This manuscript reports a 5-year cohort of newly diagnosed ALL patients and their genetic findings and outcomes, aiming to find prognostic factors and compare them to other similar reports.
Methods
Study design
This is a retrospective single-center cohort study including all patients aged from 15 years and above with newly diagnosed ALL or ambiguous lineage leukemia admitted to Instituto do Cancer do Estado de São Paulo (ICESP) from Universidade de Sao Paulo between January/2015 and December/2020. The local institutional review board approved this study. Diagnosis of ALL was based on the WHO criteria through morphology and immunophenotyping methods [Citation13]. Patients with a previous diagnosis of chronic myeloid leukemia and lymphoblastic lymphoma were excluded from the analysis. Study data were collected and managed using REDCap electronic data capture tools hosted at the University of Sao Paulo [Citation14]. This study was approved by institutional review board from Faculdade de Medicina da Universidade de São Paulo (Plataforma Brasil, CAAE 80534117.4.0000.0068) and was carried out in accordance with Declaration of Helsinki for experiments involving humans. Informed consent was waived by the Ethics committee.
Genetic evaluation
All diagnostic samples were processed by the Laboratory of Tumor Biology and Cytogenetics from Hospital das Clínicas (HCFMUSP). Bone marrow (BM) or peripheral blood enriched with blasts (in ‘dry tap’ cases) were analyzed. All patients were screened for BCR-ABL1 rearrangement through FISH or RT–PCR during the corticosteroid pre-phase. In addition, all samples from patients with B-lineage ALL were evaluated for TCF3-PBX1, KMT2A-AFF4, and ETV6-RUNX1 (patients < 25 years old) fusions through RT–PCR[Citation15]. Conventional karyotyping was attempted in all samples by standardized techniques [Citation16]. In CD10-negative cases, dual-color break-apart FISH probe for 11q23 gene (KMT2A) was carried out. Patients who did not have any of these mentioned fusions detected were eligible to retrospectively screen for CRLF2 rearrangement (CRLF2-r, dual-color break-apart FISH probe, Cytocell). Fusions involving ABL1 and PDGFRB genes were also investigated by FISH (dual-color break-apart probes, Metasystems and Cytocell, respectively). Deletions encompassing IKAROS (IKZF1 gene) could be inferred from finding expression of IKZF1 dominant-negative isoforms (ik6 and ik10, resulting from exons 4–7 and 2–7 deletions, respectively) as previously reported [Citation17]. All primers followed internal validation and manufacturer’s recommendations (Invitrogen). Part of genetic experiments was performed in remnant cytogenetic pellets or RNA stored at the diagnostic laboratory. Genetic evaluation is summarized in a flowchart available in supplementary appendix.
Treatment and definitions
Over these five years, patients were treated according to our local protocol, which allocated patients to a given regimen according to age and Philadelphia chromosome status. Philadelphia-positive patients received chemotherapy plus tyrosine-kinase inhibitor (TKI), following GRAAPH-2005 [Citation18], Hyper-CVAD [Citation19], or TKI plus corticosteroids [Citation20], depending on their age and status performance. Patients were referred to alloHCT if they had not achieved major molecular remission (quantitative RT–PCR BCR-ABL1 < 0.1%) in bone marrow aspirate and they had a suitable donor as well as eligibility for the procedure. This option was discussed thoroughly with all patients, weighing the individual relapse risk against HCT-related toxicity and mortality. Philadelphia-negative patients were treated as follows: pediatric protocol BFM-based if they were young (aBFM, 2015-2017: ≤ 35 years old; 2018-2020: ≤ 50 years old) [Citation21]; adapted GRAALL-SA1 (elderly-GRAALL) if they were older (> 50 years old) [Citation22]. Before 2018, patients between 36–50 years received adapted Hyper-CVAD protocol (HCVAD) [Citation23]. Rituximab was not available for CD20-positive ALL cases as it is not reimbursed in the public health Brazilian system. In the BFM-based protocol, from 2018 onwards, native E. coli asparaginase was replaced by peg-asparaginase. Chemotherapy regimens are detailed in the supplementary appendix.
Throughout ALL treatment, the disease was monitored by BM aspirate after each cycle of induction, consolidation, and reinduction, and, afterward, every three months during maintenance. Samples were sent for measurable residual disease (MRD) assessment by flow cytometry (FC) in our local laboratory. Even though MRD was analyzed, the methodology concerning sample processing and interpretation greatly varied in the last few years, hampering a proper report on this aspect. The level of BCR-ABL1 fusion by quantitative PCR was monitored in Ph-positive subjects in our already validated laboratory [Citation24]. AlloHCT in first complete remission (CR1) for Ph-negative patients was recommended if there was a high risk cytogenetic (KMT2A rearrangement, complex karyotype [≥ 5 chromosomal abnormalities], hypodiploidy [<40 chromosomes], near-triploidy [60-78 chromosomes], CRLF2 rearrangement), an early-T cell precursor (ETP) phenotype or positive MRD after the consolidation block in BFM regimen or after the third HCVAD course. ETP cases were defined as already published [Citation25].
Definition of complete remission (CR), relapse-free survival (RFS), and overall survival (OS) also followed the international standardized response criteria [Citation26]. Early death was defined as death before induction response evaluation within 30 days after initiation of therapy. Complete remission status was assessed at the end of the first induction cycle.
Central nervous system (CNS) disease was examined through cytologic evaluation of cerebrospinal fluid (CSF) and classified as follows – CNS 1: no detectable leukemia cells in the CSF; CNS 2: less than 5 WBC/μL and a positive ‘cytospin’ for blasts; CNS 3: more than 5 WBC/μL and a positive cytospin [Citation27]. Children’s Oncology Group (COG) algorithm was used to classify CNS disease (detailed in supplementary appendix) [Citation28]. Traumatic lumbar puncture (LP) was defined as more than ten red blood cells/μL of CSF [Citation27]. Intrathecal prophylaxis and treatment for CNS involvement were performed as recommended in all protocols.
All patients received anti-infection prophylaxis with acyclovir, trimethoprim-sulfamethoxazole, and fluconazole. In GRAAPH/HCVAD protocols, quinolone prophylactic during chemotherapy nadir was also recommended.
Statistical analysis
Pairwise comparisons between patient subgroups were performed by Mann–Whitney or Kruskal–Wallis tests for continuous variables and by Pearson’s chi-square or Fisher’s exact test for categorical variables. Event-free survival (EFS) was calculated from the date of diagnosis until refractoriness, relapse, or death. Survival curves were plotted with the Kaplan-Meier method and compared using log-rank test. Median follow-up time was estimated by reversing the codes for the censoring indicator in the Kaplan-Meier analysis. Cumulative incidence of relapse (CIR) was calculated considering death as a competitor and compared by Gray’s test [Citation29]. Association between factors was investigated through logistic regression and factors associated with survival endpoints by Cox regression. Univariate (UVA) hazard estimates were generated with unadjusted Cox proportional hazards models. Covariates clinically relevant were included in the multivariate model (MVA). All analyses were done using R software package version v 3.5.1 (R Foundation for Statistical Computing; www.r-project.org) and a two-sided p-value <0.05 was considered statistically significant.
Results
Patients
A total of 104 patients were diagnosed with ALL during the study period, with a median age of 37.5 years (interquartile range [IQR], 15-79) and with a male sex majority (57.7%). Obesity (body mass index [BMI] ≥ 30 kg/m2) was found in 17.3% of subjects. Most ALL cases were of B-lineage (75%), followed by T-lineage (20.2%) and ambiguous lineage (4.8%). Increased WBC (≥ 30 × 109/L for B-lineage or ambiguous lineage and ≥ 100 × 109/L for T-cell ALL) was found in 6.7% of cases. Among T-lineage cases, the ETP subtype was seen in 30% (6/20), whereas the Philadelphia chromosome was detected in 42.3% of B-cell ALL (33/78) and 1 out of 5 cases of ambiguous lineage leukemia. Cerebrospinal fluid (CSF) assessment at the diagnosis was performed in 103/104 (one patient died before the first LP). Regarding CNS disease classification by cytology, CNS2 and CNS3 status were found in 14.6% (15/103) and 1.9% (2/103) of patients. Traumatic LP occurred in 50.5%. Overall, 15.5% (16/103) had positive CSF cytology, with 9/16 LP being traumatic. Flow cytometry of CSF was carried out in 72 cases, with positivity for ALL disease in 26.4% (19/72). CSF cytology had 53% (95% confidence interval [CI] 29-76) and 98.1% (95% CI 90-100) of sensitivity and specificity, respectively (considering FC as gold-standard for CSF assessment). Performing the first LP after the beginning of chemotherapy was associated with more traumatic LP (56 versus 31%, p = 0.019). Patients with positive CSF cytology or FC had a higher initial WBC (p = 0.007). The presence of peripheral blasts at the time of LP was also associated with CSF positivity (p = 0.014). The remaining baseline features are summarized in .
Table 1. Baseline features of patients (n = 104).
Genetic features
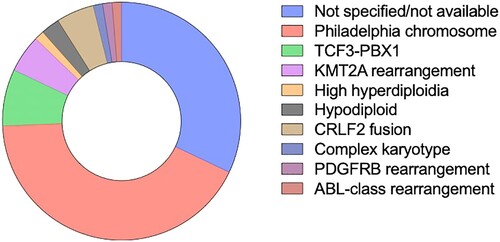
As previously mentioned, among B-lineage cases (78/104), the Philadelphia chromosome was detected in 33 cases. Remaining baseline genetic findings found were: 6 cases with t(1;19)(q23;p13) or TCF3-PBX1 fusion (7.7%), 4 KMT2A rearrangements (5.1%) [being 3/4 the t(4;11)(q21;q23) KMT2A-AFF1 fusion and 1 t(11;19)(q23;p13)], 4 CRLF2-r (5.1%) (3/4 cases with a IGH-CRLF2 fusion and 1 case with a P2RY8-CRLF2 fusion), 1 case had a hyperdiploid karyotype, 1 case with complex karyotype and 2 cases with low hypodiploidy. Additionally, 1 case with ABL1 rearrangement (1.2%), 1 case with an FGFR rearrangement, and 1 case with PDGFRB rearrangement was detected. Overall, only 24 cases had available sample for testing the presence of ik6/ik7 isoforms, of which 3/24 were positive (all of them not CRLF2 rearranged). Karyotype with no metaphases occurred in 11/104 cases. In T-cell ALL cases, karyotype was complex in 3/17 cases. Philadelphia-positive patients and those with an alleged Ph-like fusion had higher initial WBC (p = 0.06). No interaction was detected between BMI and Ph-like status (p = 0.73). D-dimer was greatly increased in KMT2A rearranged cases (mean value 28663 vs. 3135 IU/dl, p = 0.008). Genetic features of B-lineage ALL are depicted in .
Figure 1. Graphic displaying genetic baseline features of B-lineage ALL cohort.
Response and survival outcomes
Overall, two patients died from early complications during pre-phase and were excluded from survival analysis. Among Ph-negative subjects, 42 received aBFM, 8 received HCVAD, and 18 received elderly-GRAALL. Regarding Ph-positive patients, 32 received chemotherapy plus TKI (GRAAPH-2005 or HCVAD), and two received only corticosteroids plus TKI.
Out of 102 cases, an early death rate of 10.8% was found, 5/11 within BFM-inspired protocol, and four patients treated with the elderly’s regimen. Ambiguous lineage leukemia was more frequently seen in this subset of patients (18.2 vs. 2.2%, p = 0.029). Nine patients were primary refractory to the remission induction regimen (6/9 salvaged with ‘FLAG-IDA,’ 2 with ‘high-dose cytarabine plus methotrexate,’ and one was referred to palliative care). Ph-positive refractory patients received dasatinib along with chemotherapy as second-line therapy.
AlloHCT was performed in 18 cases, being ten performed in first CR. Median time for alloHCT in CR1 was 8.7 months. Eight out of 10 patients allografted in CR1 and 5/8 in CR2 remained alive during follow-up. No relapses were seen in the former group, while one relapse occurred in CR2 patients.
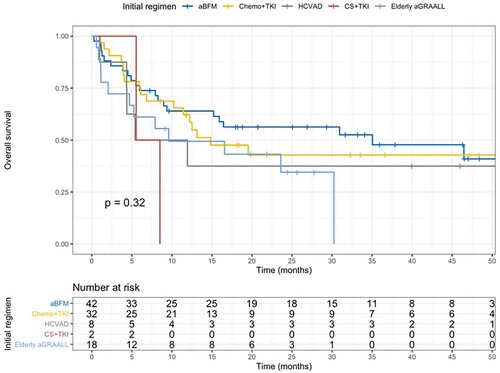
Median follow-up time was 33 months. Three-year overall survival (OS) and 3-year EFS were 42.8% (95% CI 33.7-54.5) and 30.8% (95% CI 22.2-42.8), respectively. Median OS was 16.4 months for the whole cohort. For patients treated within aBFM (Ph-negative patients under 50 years), 3-year OS and EFS were 52.5% and 36.3%, respectively. Intensively treated Philadelphia-positive patients had 42.8% and 38.6% of OS and EFS. Ph-negative elderly patients treated with aGRAALL reached 2 year-OS of 34.6% and EFS of 20.6% ().
Figure 2. OS Kaplan-Meier curve according to the initial regimen.
Univariate analysis for OS showed that age, extramedullary disease, and platelet counts at the diagnosis were statistically significant (see supplementary appendix). Multivariable Cox regression showed that extramedullary disease (hazard ratio [HR] 0.42, 95%CI 0.23-0.76, p < 0.01) and platelet counts (HR 0.99, 95% CI 0.99-0.997, p = 0.04) were independently associated with OS. Aiming to analyze the impact of certain genetic subgroups of B-lineage ALL, we divided those cases into five categories: Philadelphia-positive, fusions involving KMT2A, TCF3-PBX1 fusion, Ph-like fusion (fusions encompassing CRLF2, ABL1, FGFR1, or PDGFRB gene) and not specified. Patients with known genetic lesions such as ploidy alterations or complex karyotype were excluded from this analysis due to the low number of subjects. In , we display OS and EFS for such subsets. Outcomes for each Ph-like case are depicted in . Two out of 6 patients with ETP leukemia remained alive after alloHCT in CR1.
Figure 3. OS and EFS curves according to the genetic subset within B-lineage cases.
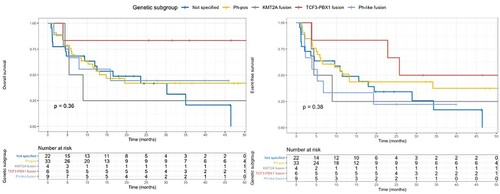
Table 2. Treatment and outcomes of patients with ‘Ph-like’ alterations.
CIR was 41.7% in 3 years (95% CI 30.7-52.2), while 3-year non-relapse mortality was 29.3% (95% CI 20.5-38.6). Univariate analysis for relapse resulted in initial WBC (HR 1.01 [95% CI 1-1.01], p = 0.03) and positive CNS disease by cytology (HR 2.16 [95% CI 1.01-4.61], p = 0.046) being significant (see supplementary appendix). None of these variables reached statistical significance in multivariable analysis for relapse. Among relapses, there was one isolated CNS relapse, 13 concurrent BM and CNS relapses, and 16 medullary relapses.
Discussion
In this manuscript, we reported our findings from a single-center cohort of adult patients with newly diagnosed ALL. Presenting features of those patients resemble previously reported cohorts, with a substantial proportion of Philadelphia-positive patients. Preliminary evidence points that Philadelphia-positive ALL, as well as Ph-like cases, are more common in Hispanic patients [Citation9]. Perez-Andre et al. have demonstrated through a genome-wide association study that GATA 3 rs3824662 risk allele was associated with Ph-like alterations and it is more frequent in Hispanics subjects [Citation30]. In our study, we were not able to screen for Ph-like expression signature as standardized by other groups [Citation31], but genetic lesions underlying the Ph-like subset were found in 8.7% of B-lineage cases. Minor subgroups are missed by using a targeted-FISH approach, as a 20-25% prevalence is estimated within B-lineage cases in other studies [Citation9]. Similar approaches to screen Ph-like cases were recently published by Mayo Clinic, which reported an incidence of 9%, similar to ours, even though more FISH probes were used [Citation32]. The UKALL group also has used a combined approach using FISH and MLPA for such cases, with 7% of B-lineage patients having a JAK-STAT or ABL1 fusion identified [Citation33]. A greater frequency of t(1;19) was also noticed in our cohort (7.7%) when compared to other groups from Europe (3-5%) [Citation33–35]. This fusion is reported to be more frequent in African children or black subjects from the United States, being that one primary reason, as Brazil has a substantial African ethnic background [Citation36,Citation37]. This is the first study to report Ph-like alterations in adults from Latin America.
In our study, no interaction between obesity and Ph-like cases was noticed, going against preliminary findings reported by Mittelman et al. in a cohort of 23 patients having a CRLF2 rearrangement [Citation38]. Stock and colleagues from the CALGB group found an impact of obesity on disease-free survival of newly diagnosed Ph-negative ALL cases, regardless of their level of CRLF2 expression [Citation39]. This finding was also reported by Greenwood et al. [Citation40] in an Australian younger cohort. Noticeably, the prevalence of obesity in this cohort was higher than that reported by our study (32 vs. 17%). Unfortunately, the small number of patients in those subsets (obese patients, CRLF2-r) impairs considerable comparisons among studies. Arguably, differences in the prevalence of obesity across distinct populations might partially explain such discrepancy in CRLF2-r incidence and response.
-Reiterating recent reports, the incorporation of TKI into the frontline regimen of Ph-positive cases can provide comparable long-term survivals when compared to the Ph-negative counterpart [Citation33]. We focused on this distinctive subset in another publication [Citation10]. Patients with TCF3-PBX1 fusion in our cohort fare better in terms of survival, which suggests its positive prognostic impact [Citation41]. Unfortunately, some patients with Ph-like fusions in this cohort were not managed as appropriate because they were tested retrospectively. Ideally, those patients should receive intensive therapy followed by alloHCT in CR1, at least in adults [Citation42]. One case with an ABL1 fusion received nilotinib after one course of chemotherapy, sustaining remission until the alloHCT, suggesting an antileukemic effect in this translocation. DenBouer et al. reported on a multicenter pediatric cohort of 122 patients with ABL-class fusions, describing a poor long-term survival and a significant role of alloHCT in survival, although partially offset by its toxicity [Citation43]. Different groups have reported benefit from associating TKI to chemotherapy in those cases, making it pivotal to find this alteration upfront adequately [Citation44,Citation45].
Publications from the developing world have pointed that those pediatric protocols are beneficial to adolescents and young adults also in this setting [Citation46]. Similar reports from Brazil have demonstrated worse long-term survivals [Citation10,Citation12,Citation47].
After analyzing our findings, we conclude that we still face excessive non-relapse mortality that significantly compromises our results. Delays in alloHCT, as well as the unavailability of new agents, also impact long-term survival. After this analysis, we have implemented this targeted-FISH approach to classify our B-lineage cases and have worked on increasing alloHCT availability and MRD standardization in our center. Measures aiming at decreasing early infection are also desirable.
Supplemental Material
Download MS Word (139.7 KB)Acknowledgements
To Division of Hematology of Hospital das Clinicas da Universidade de Sao Paulo, for all support during this study, and to all patients and their caregivers.
Disclosure statement
No potential conflict of interest was reported by the author(s).
References
- Bassan R, Bourquin J-P, DeAngelo DJ, et al. New approaches to the management of adult acute lymphoblastic leukemia. J Clin Oncol [Internet]. 2018;36:3504–3519. Available from: http://ascopubs.org/doi/10.1200/JCO.2017.77.3648
- Mitchell RJ, Kirkwood AA, Barretta E, et al. IKZF1 alterations are not associated with outcome in 498 adults with B-precursor ALL enrolled in the UKALL14 trial. Blood Adv [Internet]. 2021;5:3322–3332. Available from: https://ashpublications.org/bloodadvances/article/5/17/3322/476674/IKZF1-alterations-are-not-associated-with-outcome
- Ribera J-M, Morgades M, Ciudad J, et al. Chemotherapy or allogeneic transplantation in high-risk Philadelphia chromosome–negative adult lymphoblastic leukemia. Blood [Internet]. 2021;137:1879–1894. Available from: https://ashpublications.org/blood/article/137/14/1879/474081/Chemotherapy-or-allogeneic-transplantation-in-high
- Gu Z, Churchman ML, Roberts KG, et al. PAX5-driven subtypes of B-progenitor acute lymphoblastic leukemia. Nat Genet [Internet]. 2019;51:296–307. Available from: http://www.nature.com/articles/s41588-018-0315-5
- Iacobucci I, Kimura S, Mullighan CG. Biologic and therapeutic implications of genomic alterations in acute lymphoblastic leukemia. J Clin Med [Internet]. 2021;10:3792. Available from: https://www.mdpi.com/2077-0383/10/17/3792
- Cario G, Leoni V, Conter V, et al. BCR-ABL1-like acute lymphoblastic leukemia in childhood and targeted therapy. Haematologica [Internet]. 2020;105:2200–2204. Available from: https://new.haematologica.org/article/view/9757
- Herold T, Gökbuget N, Herold T. Philadelphia-Like acute lymphoblastic leukemia in adults. Curr Oncol Rep. 2017: 1–6.
- Bajel A, George B, Mathews V, et al. Adult ALL: treatment outcome and prognostic factors in an Indian population using a modified German ALL (GMALL) protocol. Leukemia [Internet]. 2007;21:2230–2233. Available from: http://www.nature.com/doifinder/10.1038/sj.leu.2404785
- Jain N, Roberts KG, Jabbour E, et al. Ph-like acute lymphoblastic leukemia: a high-risk subtype in adults. Blood [Internet]. 2017;129:572–581. Available from: https://ashpublications.org/blood/article/129/5/572/36151/Phlike-acute-lymphoblastic-leukemia-a-highrisk
- Silva WF, Silverio A, Duarte BKL, et al. Philadelphia-positive B-lymphoblastic leukemia in a middle-income country – A real-world multicenter cohort. Leuk Res [Internet]. 2021;110:106666. Available from: https://linkinghub.elsevier.com/retrieve/pii/S0145212621001673
- Crespo-Solis E, Espinosa-Bautista K, Alvarado-Ibarra M, et al. Retrospective study of adults With acute lymphoid leukemia in Mexico city: first report of the working group for acute leukemia (GTLA). Clin Lymphoma Myeloma Leuk [Internet]. 2017;17:S252. Available from: https://linkinghub.elsevier.com/retrieve/pii/S2152265017310182
- Fernandes da Silva Junior W, Medina AB, Yamakawa PE, et al. Treating adult acute lymphoblastic leukemia in Brazil—Increased early mortality using a German multicenter acute lymphoblastic leukemia-based regimen. Clin Lymphoma Myeloma Leuk [Internet]. 2018;18:e255–e259. Available from: http://linkinghub.elsevier.com/retrieve/pii/S2152265018301538
- Swerdlow SH, Campo E, Harris NL, et al. , WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues (Revised 4th edition). 4th ed Lyon: Lyon: IARC; 2017.
- Harris PA, Taylor R, Thielke R, et al. Research electronic data capture (REDCap)—A metadata-driven methodology and workflow process for providing translational research informatics support. J Biomed Inform [Internet]. 2009;42:377–381. Available from: http://linkinghub.elsevier.com/retrieve/pii/S1532046408001226
- van Dongen J, Macintyre E, Gabert J, et al. Standardized RT-PCR analysis of fusion gene transcripts from chromosome aberrations in acute leukemia for detection of minimal residual disease. Leukemia [Internet]. 1999;13:1901–1928. Available from: http://www.nature.com/articles/2401592
- Tanizawa RS da S, Kumeda CA, Azevedo Neto Rd, et al. Karyotypic and fluorescent in situ hybridization study of the centromere of chromosome 7 in secondary myeloid neoplasms. Rev Bras Hematol Hemoter [Internet]. 2011;33:425–431. Available from: http://www.rbhh.org/?doi=10.5581/1516-8484.20110117
- Moreira LBP, Queiróz RdP, Suazo VK, et al. Detection by a simple and cheaper methodology of Ik6 and Ik10 isoforms of the IKZF1 gene is highly associated with a poor prognosis in B-lineage paediatric acute lymphoblastic leukaemia. Br J Haematol [Internet]. 2019;187. Available from: https://onlinelibrary.wiley.com/doi/abs/10.1111/bjh.16172
- Chalandon Y, Thomas X, Hayette S, et al. Randomized study of reduced-intensity chemotherapy combined with imatinib in adults with Ph-positive acute lymphoblastic leukemia. Blood. 2015;125:3711–3719.
- Daver N, Thomas D, Ravandi F, et al. Final report of a phase II study of imatinib mesylate with hyper-CVAD for the front-line treatment of adult patients with Philadelphia chromosome-positive acute lymphoblastic leukemia. Haematologica. 2015;100:653–661.
- Foa R, Vitale A, Vignetti M, et al. Dasatinib as first-line treatment for adult patients with Philadelphia chromosome – positive acute lymphoblastic leukemia. Blood. 2011;118:6521–6528.
- Diogenes E, Junior P, Pracchia LF, et al. Prognostic factors in adolescent and adult patients With acute lymphoblastic leukemia With Two protocols of chemotherapy : A cross-sectional study. Clin lymphoma. Myeloma Leuk [Internet]. Elsevier Inc; 2015;15:e7–14. Available from: http://doi.org/10.1016/j.clml.2014.07.006
- Hunault-Berger M, Leguay T, Thomas X, et al. A randomized study of pegylated liposomal doxorubicin versus continuous-infusion doxorubicin in elderly patients with acute lymphoblastic leukemia:The GRAALL-SA1 study. Haematologica. 2011;96:245–252.
- Kantarjian HM, O’Brien S, Smith TL, et al. Results of treatment With Hyper-CVAD, a dose-intensive regimen, in Adult Acute lymphocytic leukemia. J Clin Oncol [Internet]. 2000;18:547–547. Available from: http://ascopubs.org/doi/10.1200/JCO.2000.18.3.547
- Pfeifer H, Cazzaniga G, van der Velden VHJ, et al. Standardisation and consensus guidelines for minimal residual disease assessment in Philadelphia-positive acute lymphoblastic leukemia (Ph + ALL) by real-time quantitative reverse transcriptase PCR of e1a2 BCR-ABL1. Leukemia [Internet]. 2019;33:1910–1922. Available from: http://www.nature.com/articles/s41375-019-0413-0
- Coustan-Smith E, Mullighan CG, Onciu M, et al. Early T-cell precursor leukaemia: a subtype of very high-risk acute lymphoblastic leukaemia. Lancet Oncol. 2009;10:147–156.
- NCI Dictionary of Cancer Terms [Internet]. [cited 2020 Nov 17]. Available from: https://www.cancer.gov/publications/dictionaries/cancer-terms
- Pui C-H, Howard SC. Current management and challenges of malignant disease in the CNS in paediatric leukaemia. Lancet Oncol [Internet]. 2008;9:257–268. Available from: https://linkinghub.elsevier.com/retrieve/pii/S1470204508700706
- Maloney KW, Devidas M, Wang C, et al. Outcome in Children With standard-risk B-cell acute lymphoblastic leukemia: Results of Children’s Oncology Group trial AALL0331. J Clin Oncol [Internet]. 2020;38:602–612. Available from: https://ascopubs.org/doi/10.1200/JCO.19.01086
- Scrucca L, Santucci A, Aversa F. Competing risk analysis using R: an easy guide for clinicians. Bone Marrow Transplant [Internet]. 2007;40:381–387. Available from: http://www.nature.com/articles/1705727
- Perez-Andreu V, Roberts KG, Harvey RC, et al. Inherited GATA3 variants are associated with Ph-like childhood acute lymphoblastic leukemia and risk of relapse. Nat Genet [Internet]. 2013;45:1494–1498. Available from: http://www.nature.com/articles/ng.2803
- Roberts KG, Li Y, Payne-Turner D, et al. Targetable kinase-activating lesions in Ph-like acute lymphoblastic leukemia. N Engl J Med [Internet]. 2014;371:1005–1015. Available from: http://www.nejm.org/doi/10.1056/NEJMoa1403088
- Abdel-Rahman ZH, Heckman MG, Anagnostou T, et al. Identification of adult Philadelphia-like acute lymphoblastic leukemia using a FISH-based algorithm distinguishes prognostic groups and outcomes. Blood Cancer J [Internet]. 2021;11:156. Available from: https://www.nature.com/articles/s41408-021-00538-9
- Moorman A V, Barretta E, Butler ER, et al. Prognostic impact of chromosomal abnormalities and copy number alterations in adult B-cell precursor acute lymphoblastic leukaemia: a UKALL14 study. Leukemia [Internet]. 2021. Available from: https://www.nature.com/articles/s41375-021-01448-2
- Beldjord K, Chevret S, Asnafi V, et al. Oncogenetics and minimal residual disease are independent outcome predictors in adult patients with acute lymphoblastic leukemia. Blood [Internet]. 2014;123:3739–3749. Available from: https://ashpublications.org/blood/article/123/24/3739/32968/Oncogenetics-and-minimal-residual-disease-are
- Chiaretti S, Vitale A, Cazzaniga G, et al. Clinico-biological features of 5202 patients with acute lymphoblastic leukemia enrolled in the Italian AIEOP and GIMEMA protocols and stratified in age cohorts. Haematologica [Internet]; 2013(98):1702–1710. Available from: http://www.haematologica.org/cgi/doi/10.3324/haematol.2012.080432
- Pui C-H. Results of therapy for acute lymphoblastic leukemia in black and white children. JAMA [Internet]. 2003;290:2001. Available from: http://jama.jamanetwork.com/article.aspx?doi=10.1001/jama.290.15.2001
- Vaughan J, Bouwer N, Willem P, et al. The translocation t(1;19)(q23;p13) (TCF3/PBX1 fusion) is the most common recurrent genetic abnormality detected amongst patients with B-cell lymphoblastic leukaemia in johannesburg. South Africa. South African J Oncol [Internet]. 2021;5. Available from: https://www.sajo.org.za/index.php/sajo/article/view/179
- Mittelman SD, Kim J, Raca G, et al. Increased prevalence of CRLF2 rearrangements in obesity-associated acute lymphoblastic leukemia. Blood [Internet]. 2021;138:199–202. Available from: https://ashpublications.org/blood/article/138/2/199/475790/Increased-prevalence-of-CRLF2-rearrangements-in
- Stock W, Luger SM, Advani AS, et al. A pediatric regimen for older adolescents and young adults with acute lymphoblastic leukemia: results of CALGB 10403. Blood [Internet]. 2019;133:1548–1559. Available from: http://www.bloodjournal.org/lookup/doi/10.1182/blood-2018-10-881961
- Greenwood M, Trahair T, Sutton R, et al. An MRD-stratified pediatric protocol is as deliverable in adolescents and young adults as children with ALL. Blood Adv [Internet]. 2021. Available from: https://ashpublications.org/bloodadvances/article/doi/10.1182/bloodadvances.2021005576/477382/An-MRD-stratified-pediatric-protocol-is-as
- Yilmaz M, Kantarjian HM, Toruner G, et al. Translocation t(1;19)(q23;p13) in adult acute lymphoblastic leukemia – a distinct subtype with favorable prognosis. Leuk Lymphoma [Internet]. 2021;62:224–228. Available from: https://www.tandfonline.com/doi/full/10.1080/10428194.2020.1824071
- Frisch A, Ofran Y. How i diagnose and manage Philadelphia chromosome-like acute lymphoblastic leukemia. Haematologica [Internet]. 2019;104:2135–2143. Available from: http://www.haematologica.org/lookup/doi/10.3324/haematol.2018.207506
- den Boer ML, Cario G, Moorman AV, et al. Outcomes of paediatric patients with B-cell acute lymphocytic leukaemia with ABL-class fusion in the pre-tyrosine-kinase inhibitor era: a multicentre, retrospective, cohort study. Lancet Haematol [Internet]. 2021;8:e55–e66. Available from: https://linkinghub.elsevier.com/retrieve/pii/S2352302620303537
- Moorman AV, Schwab C, Winterman E, et al. Adjuvant tyrosine kinase inhibitor therapy improves outcome for children and adolescents with acute lymphoblastic leukaemia who have an ABL-class fusion. Br J Haematol [Internet]. 2020;191:844–851. Available from: https://onlinelibrary.wiley.com/doi/10.1111/bjh.17093
- Tanasi I, Ba I, Sirvent N, et al. Efficacy of tyrosine kinase inhibitors in Ph-like acute lymphoblastic leukemia harboring ABL-class rearrangements. Blood [Internet]. 2019;134:1351–1355. Available from: https://ashpublications.org/blood/article/134/16/1351/374966/Efficacy-of-tyrosine-kinase-inhibitors-in-Phlike
- Almanza-Huante E, Espinosa-Bautista K, Rangel-Patiño J, et al. Comparison of Two pediatric-inspired regimens to Hyper-CVAD in Hispanic adolescents and young Adults With Acute lymphoblastic leukemia. Clin Lymphoma Myeloma Leuk [Internet]. 2021;21:55–62.e2. Available from: https://linkinghub.elsevier.com/retrieve/pii/S2152265020303797
- Gurgel LA, Oliveira Ds, Leitão JPV, et al. Overall survival in patients with acute lymphoblastic leukemia treated with CALGB 8811 protocol in low income country. J BONE MARROW Transplant Cell Ther [Internet]. 2021;2:95. Available from: https://www.jbmtct.com.br/seer/index.php/jbmtct/article/view/95