
ABSTRACT
Purpose
Pediatric acute myeloid leukemia (AML) with KMT2A rearrangements has a very different prognosis. Poor outcomes cannot be avoided even after hematopoietic stem cell transplantation. In order to investigate the prognosis and efficacy, we conducted a retrospective analysis.
Patients and methods
We retrospectively analyzed a total of 32 children with KMT2A rearrangements AML treated in our hospital between January 2015 and February 2021.
Results
The proportion of patients with KMT2A-rearranged in the medium-risk group of overall survival (OS) and event-free survival (EFS) was 100%. No differences in OS, EFS and cumulative incidence of relapse (CIR) were detected between the haploidentical hematopoietic stem cell transplantation (haplo-HSCT) and full matched HSCT (P = 0.289, P = 0.303, P = 0.303). Acute graft-versus-host disease (aGVHD) was often detected in the haplo-HSCT cohort, while full matched HSCT had no obvious aGVHD, assessed as≤1 grade (P < 0.05). Patients in the medium-risk pediatric group could acquire 100% OS and EFS only after chemotherapy. There was no significant difference in OS, EFS and CIR between full matched HSCT and haploidentical transplantation in pediatric AML with KMT2A rearrangements, but full matched HSCT seemed to have a lower death rate. The severity of aGVHD in the full matched HSCT was less than that in the haploidentical transplantation group.
Conclusion
The primary choice of donor can be HLA-matched sibling donors or matched unrelated donors for children with AML with KMT2A rearrangements, and the secondary choice can be haploid donors.
Introduction
Acute myeloid leukemia (AML) accounts for about 15%–20% of childhood leukemias, and its incidence in the pediatric population is much lower than acute lymphocytic leukemia (ALL), which accounts for 70%–80% [Citation1, Citation2]. Although the survival rate of AML has been greatly improved, the overall survival (OS) rate of AML children is approximately 60%–70%, which is still lower than that of ALL children [Citation3]. Pediatric AML is a heterogeneous disease, showing variability in morphology, cytogenetic abnormalities and treatment response [Citation4]. The prognostic investigation of AML and various chromosomal abnormalities is extremely important for determining treatment strategies. The mixed lineage leukemia (MLL) gene, which is now known as the lysine-specific methyltransferase 2A (KMT2A) gene, located at chromosome band 11q23, has been detected in 15%–20% of pediatric acute myeloid leukemia cases with different partner chromosomes [Citation5]. Lysine methyltransferase 2a encoded by KMT2A regulates gene expression via methylation of histone residues and epigenetic modifications, which plays a critical role in normal embryonic development and hematopoiesis [Citation6]. Translocations involving KMT2A result in KMT2A fusion proteins that contribute to aberrant expression of downstream mediators [Citation7]. These fusion proteins may cause changes in the epigenetic regulation of hematopoietic cells, leading to differentiation arrest and promoting leukemia. KMT2A rearrangements are more common in children than adults with a particularly high occurrence in infant AML. The fusion partner profile is also unique to each age group [Citation8]. While studies in children have shown that KMT2A rearrangements are clearly associated with inferior outcomes with an event-free survival (EFS) of 44% and an OS of 56% along with higher rates of early death and relapse, the true prognostic value is highly dependent on the fusion partner gene. More than 100 different fusion partners have been identified. The KMT2A/MLLT3 fusion resulting from t(9;11)(p22;q23) is the most common KMT2A rearrangement in children. The prognosis of children harboring t(9;11)(p22;q23) remains controversial. Owing to the low frequency of most KMT2A fusions, not all rearrangements have well-defined outcomes [Citation9–11]. This study retrospectively investigated the outcomes and prognostic factors of pediatric AML patients with KMT2A rearrangements who were treated at our institution over a 5-year period.
Materials and methods
Patients
Between January 2015 and February 2021, a total of 32 children with KMT2A rearrangements AML treated at our institution were included in this study. AML was diagnosed in accordance with MICM criteria [Citation12]. KMT2A rearrangements were detected through (i) reverse transcription polymerase chain reaction (RT–PCR) for the KMT2A gene; (ⅱ) karyotype analysis of chromosomes with classic 11q23 translocations; and/or (iii) fluorescence in situ hybridization (FISH) with split-signal KMT2A probes. Except for 4 children who stopped treatment at the beginning of therapy, the remaining patients had received induction therapy and several rounds of consolidation therapy, and then 21 children opted for allogeneic hematopoietic stem cell transplantation (allo-HSCT). The chemotherapy regimen was the Chinese Children’s Leukemia Group-acute myeloid leukemia 2015 protocol (CCLG-AML-2015), derived from the United Kingdom Medical Research Council AML (MRC-AML) protocol [Citation13]. The patients were monitored and evaluated before and after stem cell transplantation, with follow-up ending on 30 April 2021. This study was approved by the Institutional Ethics Committee of Beijing Children’s Hospital. Informed consent was obtained from the patients’ parents or guardians. All donors provided written informed consent for the protocol.
Allogeneic hematopoietic stem cell transplantation
After induction therapy and several rounds of consolidation therapy, the vast majority of patients achieved their first complete remission (CR1). Depending on chemotherapeutic efficacy, fusion gene clearance, donor availability and economic considerations, some patients received allo-HSCT in accordance with the guardians’ request.
In addition to 4 patients (receiving a matched sibling donor transplant [MSDT]), 17 children underwent haploid hematopoietic stem cell transplantation (haplo-HSCT). The preconditioning protocol was performed in accordance with previous reports [Citation14–16]. The conditioning regimen for haplo-HSCT included cytarabine (4 g/m2/d) from days−10 to−9; busulfan (3.2–4.8 mg/kg/d) from days−8 to−6; cyclophosphamide (1.8 g/m2/d) from days−5 to −4; methyl chloride hexamethylene urea nitrate (Me-CCNU) (250 mg/kg/d) on day−3; anti-thymocyte globulin (ATG) (2.5 mg/kg/d; Genzyme Polyclonals S.A.S) from days −5 to −2. Patients in the MSDT group received cytarabine (4 g/m2 per day) on day −9; busulfan (4.8 mg/kg/d) from days−8to−6; cyclophosphamide (50 mg/kg/d) from days−5to−2. Supportive treatment was administered as previously described. Patients who received MSDT regimen accepted cyclosporin A and short-term methotrexate to prevent graft versus-host disease (GVHD), and children treated with haplo-HSCT regimen received cyclosporin A, mycophenolate mofetil and short-term methotrexate to prevent GVHD [Citation17].
Definitions and assessments
A complete remission was defined as < 5% bone marrow (BM) blasts without extramedullary (EM) disease; partial response was defined as the presence of ≥5% and <20% BM blasts. Relapse was defined as morphologic evidence of disease in the marrow (recurrence of ≥5% lymphoblasts) and/or EM sites. OS was defined as the time from diagnosis to death or the final follow-up examination. EFS was defined as the time from diagnosis to the first event (remission failure, relapse or secondary malignancy) or the final follow-up examination. The cumulative incidence of relapse (CIR) was defined as the time from CR1 to the first relapse or the final follow-up examination [Citation14]. GVHD was classified according to the widely accepted National Institutes of Health consensus criteria [Citation18, Citation19].
Statistical analysis
Between-group comparisons were performed using Student’s t-test or Fisher’s exact test. Survival analysis was performed using the Kaplan-Meier method and graphs created by Prism 8.0 (Graph Pad, USA). Other analyses were primarily conducted using SPSS 24.0 (IBM, USA). P < 0.05 indicated statistical significance.
Results
Between January 2015 and February 2021, a total of 32 (21 male, 11 female) pediatric patients with t(v;11q23)/KMT2A rearrangements treated at our institution were included in this study, of whom 21 received allogeneic HSCT. Age at the study group ranged from 0.8 to 15.2 years (median, 4.4 years; average, 6.0 years). Up to the time of the last follow-up (30 April 2021), the average follow-up duration was 24.6 months (range, 1–73.8 months). To identify KMT2A rearrangements, we utilized RT–PCR detection or FISH with split-signal KMT2A probes. The 32 patients showed the following KMT2A rearrangements: KMT2A/ MLLT3 or t(9,11) in 10 patients (31.25%); KMT2A/MLLT10 or t(10,11) in 8 patients (25.00%); KMT2A/MLLT11 in 2 patients; KMT2A/PTD in 2 patients; KMT2A/ AFF1 in 1 patient; KMT2A/MLLT4 in 1 patient; KMT2A/MLLT6 in 1 patient; KMT2A/SEPT6 in 1 patient; KMT2A/MLLT1 in 1 patient; KMT2A/ELL in 1 patient, and other KMT2A rearrangements in 4 patients. According to the CCLG-AML-2015, 3 patients in our study were classified as medium-risk group, while 27 children with poor genetic marker or chemotherapeutic insensitivity were identified as being in the high-risk group. Except for the above patients, there were 2 patients without risk classification (just completing the diagnosis in hospital), and the treatment was abandoned. One patient died three months after discharge, the other patient’s parents refused to follow up. After the 3 children in the medium-risk group underwent induction chemotherapy, consolidation therapy, and maintenance treatment, as of the follow-up date, all of them have survived disease-free and could maintain a normal life. One child in the high-risk group was lost to follow-up. Five children in the high-risk group died at different stages of chemotherapy and had not yet undergone hematopoietic stem cell transplantation (the average time between diagnosis and death was 3.5 months). The details of the five patients were as follows: two children gave up treatment during the induction phase due to economic reasons and the parents’ pessimistic attitude towards the prognosis of the disease, and the patients died soon after being discharged from the hospital. One child (the fusion gene is KMT2A/MLLT10) who had hyperleukocytosis and extramedullary infiltration (multiple bone destruction, liver and spleen, and ileocecal tumor cell invasion) died during the period of subtraction treatment. Due to the neoplastic cells’ rapid infiltrating growth and blood hypercoagulability, the patient died of disseminated intravascular coagulation (DIC), acute renal failure, acute respiratory failure (type Ⅱ). The other two children all relapsed after CR1. One patient achieved CR1 after the third round of consolidation chemotherapy, however, at the same time, severe bone marrow suppression and sepsis were observed, and the patient gave up chemotherapy and refused to undergo transplantation due to poverty. Two months after discharge, the child's bone marrow recurred, and died quickly without targeted treatment. Another patient had myeloid sarcoma at the beginning of the disease. After several rounds of chemotherapy, the child's condition was relieved. However, on the eve of the preparation for transplantation, her condition had relapsed. the bone marrow blasts were 49% and ocular ultrasound revealed recurrence of extramedullary invasion in bilateral orbital lacrimal gland areas. Despite receiving a FLAG recurrence induction intensive chemotherapy regimen (fludarabine (25 mg/m2/d) from days 1–5; cytarabine (2 g/m2/d) from days 1–5; G-CSF (5ug/kg/d) from days 0–6), re-examination of BM did not remission after chemotherapy. The patient's primary disease was highly malignant and difficult to treat, and to make matters worse, the risk of transplantation is significantly increased in the patient with cardiac insufficiency and atelectasis. The parents of the child subsequently gave up therapy and the child died 10 days later. Ultimately, 21 patients in high-risk group received allo-HSCT (four HLA-MSDT and seventeen haplo-HSCT), 20 of whom received allo-HSCT at CR1. The average interval between CR1 and HSCT was 2.4 months (range, 1.3–3.7 months). Only 1 patient who experienced relapse opted for allogeneic HSCT after CR2 for further treatment. Characteristics of transplant patients are summarized in .
Table 1. Main characteristics of the 21 AML pediatric patients with t (v; 11q23)/KMT2A rearrangents.
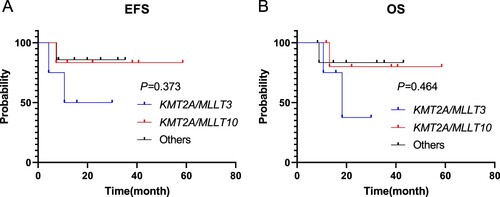
As of the follow-up date, all children who gave up treatment at the initial stage of therapy or refused hematopoietic stem cell transplantation after recurrence died excluding patients who were lost to follow-up. Patients with KMT2A-rearranged in the medium-risk pediatric group returned to normal life after induction chemotherapy, consolidation treatment and maintenance therapy, the rates of OS and EFS were 100% (). Four KMT2A-rearranged pediatric AML patients died in our cohort among 21 high-risk children underwent allogeneic hematopoietic stem cell transplantation. Among the patients who died, three patients died after receiving HSCT at CR1 (one patient owing transplant-related complications and two owing to relapse after haplo-HSCT), 1 patient died after receiving HSCT at CR2. Details of relapse after haplo-HSCT were as follows: BM relapse (2 patients); EM relapse (1 patient). One patient died of pleomorphic post-transplantation lymphoproliferative diseases (PTLD), severe interstitial pneumonia, and multi-organ failure. The differences in the OS, EFS and CIR rates between the high-risk group and the medium-risk group were shown in (P = 0.398, P = 0.428, P = 0.428, respectively). The differences in OS, EFS and CIR rates of children with full matched HSCT and haplo-HSCT in the allogeneic hematopoietic stem cell transplantation group were shown in . The OS rate of the patients with haplo-HSCT was not significantly poorer than that of children received full matched HSCT (P = 0.289), and the differences in EFS and CIR rates were likewise not detected between the two groups (P = 0.303; P = 0.303). Among the 17 children who received haplo-HSCT, they were divided into 3 groups according to different fusion genes, KMT2A/MLLT3, KMT2A/MLLT10 and Others (if the number of fusion gene case was 1, divided into Others group). The number of cases in the three groups was 4, 6, and 7, respectively. EFS and OS of the three groups were shown in (P = 0.373; P = 0.464).
Figure 1. The differences of the EFS, OS and CIR rates between the high-risk group and the medium-risk group were shown (P = 0.428, P = 0.398, P = 0.428).
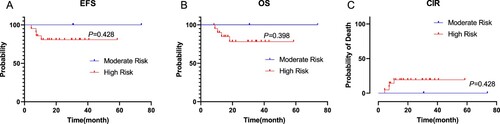
Figure 2. The differences in EFS, OS and CIR rates of children with full matched HSCT and haplo-HSCT were shown (P = 0.303, P = 0.289, P = 0.303).
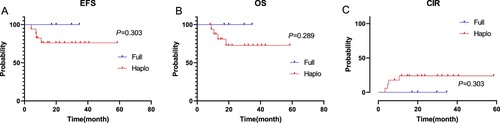
Figure 3. The differences in EFS, OS of children with KMT2A/MLLT3, KMT2A/MLLT10 and Others were shown (P = 0.373, P = 0.464).
After transplantation, children in the high-risk group all underwent a process of immune reconstitution. In children with full matched HSCT, the average time of leukocyte implantation was 12.0 days (range, 11–15 days), and the average time of platelet implantation was 12.25 days (range, 11–13 days). Correspondingly, in children who received haploidentical transplantation, the implantation time of leukocyte and platelet was 12.4 days (range, 10–19 days) and 14.0 days (range, 7–29 days) respectively (P > 0.05). Evaluation on GVHD, acute GVHD grade 2–4 was detected in 13 patients (76.4%) in the haplo-HSCT cohort, however, children with full matched HSCT had no obvious aGVHD, all assessed as ≤1 grade (P < 0.05).
Discussion
Over 100 different fusion partners have been identified in KMT2A rearrangements; however the true prognostic value is highly dependent on the fusion partner gene [Citation10,Citation11,Citation20], and the KMT2A/MLLT3 fusion resulting from t(9;11)(p22;q23) is the most common KMT2A rearrangement in children. In our retrospective study, 31.25% of children carried the KMT2A/MLLT3 fusion gene, and its proportion was roughly the same as that of previous research literature. Previous reports have indicated that the outcomes of pediatric AML with KMT2A rearrangements differ significantly according to translocation partners. Some reports have demonstrated that AML with t(9,11) was associated with better prognosis, and t(6;11)(q27;q23) and t(10;11)(p12;q23) were associated with the worst outcomes with a 5-year OS of approximately 30% [Citation9,Citation21,Citation22]. Japanese research found that, although patients with t (9,11) were the most frequently encountered, prognosis did not differ significantly from that of other 11q23 abnormalities [Citation23]. There may be various reasons for this phenomenon. The prognosis of AML children with KMT2A/MLLT3 is still controversial, and more research is needed to support it. There was only one patient with t (6,11) in our study, he remained alive in CR2, however, it needs to be added that the child had undergone a second haplo-HSCT because of molecular recurrence. This may indicate a worse prognosis for KMT2A/MLLT4 or t (6,11).
There are many reports on the stratified treatment of ALL children with KMT2A rearrangements [Citation24]. The Japanese Pediatric Leukemia/Lymphoma Study Group (JPLSG) planned a nationwide clinical trial to improve the outcomes of infants with KMT2A-r ALL; they found that risk stratification and introduction of intensive chemotherapy were effective and were able to eliminate HSCT for a subset of infants with KMT2A-r ALL [Citation25]. In this study, not all children with AML with KMT2A rearrangements required hematopoietic stem cell transplantation and could be treated with stratification. Patients assessed as medium-risk can achieve long-term disease-free survival after standard treatment with CCLG-AML2015 chemotherapy regimen, and transplantation is not required for the time being. Perhaps researchers carefully rate the risk and adopt a stratified treatment strategy based on the MICM classification and the response to chemotherapy, which will benefit the children more and avoid overtreatment.
Past research reports that the OS and EFS rates of adult AML patients with KMT2A rearrangements were 36.72%–56% and 29.33%–51% respectively [Citation26,Citation27]. The OS rate of KMT2A-rearranged pediatric AML was 52.1% and the EFS was 46.7% respectively, which is same trend as our research results. A national retrospective study about pediatric acute myeloid leukemia patients with KMT2A rearrangements in Japan found that the cumulative incidence of relapse at 5 years in patients who received HLA full matched HSCT was 25.0%, while that with haploidentical HSCT was 22.7% (P = 0.496), and no difference in leukemia-related mortality was evident [Citation23]. In our retrospective study, the rates of OS, EFS were identically no significant difference between the patients who received haplo-HSCT and full matched HSCT. In previous research, allogeneic hematopoietic stem cell transplantation (allo-HSCT), especially from HLA-matched sibling donors (MSD) or matched unrelated donors (MUD), is one of the standard options and possibly the preferred option for individuals with high-risk AML [Citation28]. Recently, haplo-HSCT was confirmed as an equally good alternative to MSD-HSCT as a post remission therapy for patients with AML in the first morphologic complete remission (CR1), who lack a matching donor [Citation29–33]. In this study, we analyzed the differences in transplant-related complications between haplo-HSCT and full matched HSCT patients. We found that a significant difference was seen in acute GVHD, and haplo-HSCT was worse than full matched HSCT (P < 0.05). This seems to suggest that full matched HSCT is better than haplo-HSCT. Although the number of patients was small, we analyzed the data of patients received full matched HSCT treatment, All 4 children had no myeloid sarcoma or hyperleukocytoemia (P = 0.004) at the beginning of the disease. They received transplantation treatment after CR1, and the KMT2A fusion gene was negative before transplantation. It reminds us that when the child meets the above conditions and has a matched sibling donor, full matched HSCT may be better than haplo-transplantation.
The haploidentical transplantation may cause hypersensitivity, cytokine release syndrome (CRS) [Citation34], grievous GVHD (grade 3–4) and severe fatal infection due to prolonged bone marrow emptying period. Patients with grade 3–4 GVHD have adverse reactions, such as poor stem cell implantation and thrombotic microangiopathy (TMA) [Citation35, Citation36].
The children in this group were treated using the Beijing protocol for myeloablative treatment, and there were no deaths related to myeloablation. Although the number and severity of acute GVHD cases were higher than those in full-matched transplant patients, only 1 haploidentical transplant patient died of aGVHD-related complications (pulmonary rejection). Furthermore, recent studies have found that the development of GVHD prophylaxis using post-transplant cyclophosphamide has effectively reduced transplant-related mortality caused by GVHD, graft rejection, and other related complications [Citation36]. A basic study showed that the anti-tumor effect of haploidentical allogeneic hematopoietic stem cell transplantation was better than that of the complete agreement in the mouse model of KMT2A gene mutation [Citation37]. Therefore, for children with high-risk AML carrying KMT2A gene mutations can accept haploidentical allogeneic hematopoietic stem cell transplantation on the basis of assessing the function of the children's various organs when lacking of a suitable fully matched donor.
Conclusion
For children with AML with KMT2A rearrangements, there was no significant difference in the recurrence rate between full matched HSCT and haploidentical transplantation, and the severity of a GVHD of the full matched HSCT was less than haplo-HSCT. Regarding the choice of donors, HLA-matched sibling donors or matched unrelated donors could be the primary choice, while the haploid donors are the secondary choice.
Acknowledgements
We would like to thank all the participating pediatricians and patients involved in the study.
Disclosure statement
No potential conflict of interest was reported by the author(s).
References
- Siegel DA, Henley SJ, Li J, et al. Rates and trends of pediatric acute lymphoblastic leukemia – United States, 2001–2014. MMWR Morb Mortal Wkly Rep. 2017;66(36):950–954.
- Horibe K, Saito AM, Takimoto T, et al. Incidence and survival rates of hematological malignancies in Japanese children and adolescents (2006–2010): based on registry data from the Japanese Society of pediatric hematology. Int J Hematol. 2013;98(1):74–88.
- Gamis AS, Alonzo TA, Perentesis JP, et al. Children's oncology group's 2013 blueprint for research: acute myeloid leukemia. Pediatr Blood Cancer. 2013;60(6):964–971.
- Balgobind BV, Zwaan CM, Pieters R, et al. The heterogeneity of pediatric MLL-rearranged acute myeloid leukemia. Leukemia. 2011;25(8):1239–1248.
- Balgobind BV, Van den Heuvel-Eibrink MM, De Menezes RX, et al. Evaluation of gene expression signatures predictive of cytogenetic and molecular subtypes of pediatric acute myeloid leukemia. Haematologica. 2011;96(2):221–230.
- Liedtke M, Cleary ML. Therapeutic targeting of MLL. Blood. 2009;113(24):6061–6068.
- Milne TA, Kim J, Wang GG, et al. Multiple interactions recruit MLL1 and MLL1 fusion proteins to the HOXA9 locus in leukemogenesis. Mol Cell. 2010;38(6):853–863.
- Meyer C, Burmeister T, Groger D, et al. The MLL recombinome of acute leukemias in 2017. Leukemia. 2018;32(2):273–284.
- Balgobind BV, Raimondi SC, Harbott J, et al. Novel prognostic subgroups in childhood 11q23/MLL-rearranged acute myeloid leukemia: results of an international retrospective study. Blood. 2009;114(12):2489–2496.
- von Neuhoff C, Reinhardt D, Sander A, et al. Prognostic impact of specific chromosomal aberrations in a large group of pediatric patients with acute myeloid leukemia treated uniformly according to trial AML-BFM 98. J Clin Oncol. 2010;28(16):2682–2689.
- Conneely SE, Stevens AM. Acute myeloid leukemia in children: emerging paradigms in genetics and new approaches to therapy. Curr Oncol Rep. 2021;23(2):16.
- Jaffe ES, Harris NL, Diebold J, et al. World Health Organization classification of neoplastic diseases of the hematopoietic and lymphoid tissues. A progress report. Am J Clin Pathol. 1999;111(1 Suppl 1):S8–12.
- Burnett AK, Russell NH, Hills RK, et al. Optimization of chemotherapy for younger patients with acute myeloid leukemia: results of the medical research council AML15 trial. J Clin Oncol. 2013;31(27):3360–3368.
- Bai L, Cheng YF, Lu AD, et al. Prognosis of haploidentical hematopoietic stem cell transplantation in non-infant children with t(v;11q23)/MLL-rearranged B-cell acute lymphoblastic leukemia. Leuk Res. 2020;91:106333.
- Xue YJ, Cheng YF, Lu AD, et al. Allogeneic hematopoietic stem cell transplantation, especially haploidentical, may improve long-term survival for high-risk pediatric patients with Philadelphia chromosome-positive acute lymphoblastic leukemia in the tyrosine kinase inhibitor era. Biol Blood Marrow Transplant. 2019;25(8):1611–1620.
- Chen H, Liu KY, Xu LP, et al. Haploidentical hematopoietic stem cell transplantation for pediatric Philadelphia chromosome-positive acute lymphoblastic leukemia in the imatinib era. Leuk Res. 2017;59:136–141.
- Huang XJ, Liu DH, Liu KY, et al. Haploidentical hematopoietic stem cell transplantation without in vitro T-cell depletion for the treatment of hematological malignancies. Bone Marrow Transplant. 2006;38(4):291–297.
- Jagasia MH, Greinix HT, Arora M, et al. National Institutes of Health consensus development project on criteria for clinical trials in chronic graft-versus-host disease: I. The 2014 diagnosis and staging working group report. Biol Blood Marrow Transplant. 2015;21(3):389–401 e1.
- Aladag E, Kelkitli E, Goker H. Acute graft-versus-host disease: a brief review. Turk J Haematol. 2020;37(1):1–4.
- Harrison CJ, Hills RK, Moorman AV, et al. Cytogenetics of childhood acute myeloid leukemia: United Kingdom Medical Research Council treatment trials AML 10 and 12. J Clin Oncol. 2010;28(16):2674–2681.
- Armand P, Kim HT, Zhang MJ, et al. Classifying cytogenetics in patients with acute myelogenous leukemia in complete remission undergoing allogeneic transplantation: a center for international blood and marrow transplant research study. Biol Blood Marrow Transplant. 2012;18(2):280–288.
- Shiba N, Yoshida K, Hara Y, et al. Transcriptome analysis offers a comprehensive illustration of the genetic background of pediatric acute myeloid leukemia. Blood Adv. 2019;3(20):3157–3169.
- Miyamura T, Kudo K, Tabuchi K, et al. Hematopoietic stem cell transplantation for pediatric acute myeloid leukemia patients with KMT2A rearrangement; a nationwide retrospective analysis in Japan. Leuk Res. 2019;87:106263.
- El Chaer F, Keng M, Ballen KK. MLL-rearranged acute lymphoblastic leukemia. Curr Hematol Malig Rep. 2020;15(2):83–89.
- Tomizawa D, Miyamura T, Imamura T, et al. A risk-stratified therapy for infants with acute lymphoblastic leukemia: a report from the JPLSG MLL-10 trial. Blood. 2020;136(16):1813–1823.
- Pigneux A, Labopin M, Maertens J, et al. Outcome of allogeneic hematopoietic stem-cell transplantation for adult patients with AML and 11q23/MLL rearrangement (MLL-r AML). Leukemia. 2015;29(12):2375–2381.
- Wang T, Zhao Y, Zhang QQ, et al. [Clinical characteristics and prognostic influencing factors of adult AML patients with MLL rearrangement]. Zhongguo Shi Yan Xue Ye Xue Za Zhi. 2020;28(3):775–780.
- Lv M, Wang Y, Chang YJ, et al. Myeloablative haploidentical transplantation is superior to chemotherapy for patients with intermediate-risk acute myelogenous leukemia in first complete remission. Clin Cancer Res. 2019;25(6):1737–1748.
- Cornelissen JJ, Blaise D. Hematopoietic stem cell transplantation for patients with AML in first complete remission. Blood. 2016;127(1):62–70.
- Sureda A, Bader P, Cesaro S, et al. Indications for allo- and auto-SCT for haematological diseases, solid tumours and immune disorders: current practice in Europe, 2015. Bone Marrow Transplant. 2015;50(8):1037–1056.
- Majhail NS, Farnia SH, Carpenter PA, et al. Indications for autologous and allogeneic hematopoietic cell transplantation: guidelines from the American Society for blood and marrow transplantation. Biol Blood Marrow Transplant. 2015;21(11):1863–1869.
- Burnett AK, Goldstone A, Hills RK, et al. Curability of patients with acute myeloid leukemia who did not undergo transplantation in first remission. J Clin Oncol. 2013;31(10):1293–1301.
- Koreth J, Schlenk R, Kopecky KJ, et al. Allogeneic stem cell transplantation for acute myeloid leukemia in first complete remission: systematic review and meta-analysis of prospective clinical trials. JAMA. 2009;301(22):2349–2361.
- Imus PH, Blackford AL, Bettinotti M, et al. Severe cytokine release syndrome after haploidentical peripheral Blood stem cell transplantation. Biol Blood Marrow Transplant. 2019;25(12):2431–2437.
- Jodele S, Dandoy CE, Myers KC, et al. New approaches in the diagnosis, pathophysiology, and treatment of pediatric hematopoietic stem cell transplantation-associated thrombotic microangiopathy. Transfus Apher Sci. 2016;54(2):181–190.
- Watanabe M, Kanda J. Recent progress in haploidentical transplantation: is this the optimal choice for alternative donor transplantation? Curr Opin Hematol. 2019;26(6):406–412.
- Guo H, Chang YJ, Hong Y, et al. Dynamic immune profiling identifies the stronger graft-versus-leukemia (GVL) effects with haploidentical allografts compared to HLA-matched stem cell transplantation. Cell Mol Immunol. 2021;18(5):1172–1185.