
ABSTRACT
Introduction
The aim of the study was to determine molecular genetic and clinical characterization of acute myeloid leukemia (AML) with trisomy 8 as the sole chromosome abnormality, a recurrent but rare chromosomal abnormality in AML.
Methods
Interphase fluorescence in situ hybridization, reverse transcriptase-quantitative polymerase chain reaction for gene rearrangement and next-generation sequencing (NGS) were performed on sole trisomy 8 AML patients.
Results
A total of 35 AML patients with trisomy 8 as the sole chromosome abnormality were screened. The most frequently mutated genes were DNMT3A(37.1%), RUNX1(28.6%), FLT3-ITD(28.6%), IDH2(22.9%), NPM1(17.1%), and ASXL1 (14.3%). The sole +8 AML patients exhibited more mutations in RUNX1 (28.6% vs. 4.8%, P = 0.001) and ASXL1 (14.3% vs. 4.8%, P = 0.039) by comparing with normal karyotype AML (NK AML) patients(n = 63). The sole +8 AML patients(n = 35) with RUNX1 or IDH2 mutations showed significantly lower WBC counts, while FLT3-ITD showed higher white blood cell (WBC) counts as compared to the corresponding wild-type groups. Total of 45.7% patients achieved complete remission (CR) after the first induction therapy. The CR rate of patients with FLT3-ITD or IDH1 mutation was significantly lower than that in the corresponding wild-type cases (P = 0.047, 0.005, respectively). The median overall survival (OS) and disease-free survival (PFS) were 18.0 (95% CI: 10.8–25.2) and 10 (95% CI: 6.7–13.3) months, respectively. FLT3-ITD mutations and allogeneic hematopoietic stem cell transplantation (allo-HSCT) were independent prognostic markers for OS in multivariable analysis.
Conclusion
The results suggest a possible association between trisomy 8 and additional mutations that may influence clinical feature and prognosis.
Introduction
Acute myeloid leukemia (AML) is an aggressive and very heterogeneous disease, characterized by bone marrow infiltration of immature leukemia cells, the wide range of chromosomal, epigenetic, and other genetic alterations [Citation1–3]. AML cases with a single trisomy constitute a heterogenous subgroup with regard to clinical, morphological, and immunophenotypic features, as well as the mutational profiles [Citation4]. Among the chromosomal alteration, trisomy 8 is the most common cytogenetic abnormality in Chinese patients with myelodysplastic syndromes(MDS), occurring at a frequency between 19.12% and 31% [Citation5,Citation6]. The aberration also can occur in other myeloid malignancies, such as chronic myelomonocytic leukemia (CMML) [Citation7], chronic myeloid leukemia (CML) [Citation8], Philadelphia chromosome negative myeloproliferative neoplasia (MPN) [Citation9], and AML [Citation10,Citation11].
Trisomy 8 could exist as a sole chromosomal aberration or one of several chromosomal changes in AML. Trisomy 8 is often seen together with translocations, including t(8;21)(q22;q22), inv(16)/t(16;16)(p13;q22), t(3;21)(q26;q22), t(15;17)(q22;q21), t(16;21)(q24;q22) [Citation10–13]. It is also found in rearrangement involving KMT2A [Citation14]. However, sole trisomy 8 is relatively rare in AML, occurring at a frequency between 3.78% and 5% [Citation4,Citation15,Citation16]. In various instances, molecular studies of these abnormalities identify specific genes implicated in the process of leukemogenesis and are now used in combination with cytogenetics as diagnostic and prognostic markers, which help to guide the clinicians in selecting the most effective therapies [Citation10]. The few available studies suggest that sole trisomy 8 AML is molecularly heterogeneous, and the clinical impact of mutations remains to be established [Citation17]. We report herein the molecular and clinical characterization of 35 adults de novo AML with trisomy 8 as the sole chromosome abnormality.
Patients and methods
Patient cohort
We reviewed the medical records of 1206 consecutive adults with de novo AML who had cytogenetic studies from January 2015 to May 2021 at the four medical institutions of hematology (Affiliated Changzhou Second Hospital of Nanjing Medical University, Wuxi Third people’s hospital, The First Affiliated Hospital of NanJing Medical University and The First Affiliated Hospital of Soochow University). 1206 AML cases were diagnosed with de novo AML according to French-American-British (FAB) classification and World Health Organization (WHO) 2016 classification [Citation18]. In 35 patients (2.9%), +8 was the sole abnormality. We retrospectively selected 187 AML patients with complete data as comparator groups: ① 63 AML patients with normal karyotype(NK AML) ② 83 AML patients with t (8;21)/RUNX1-RUNX1T1, and ③ 41 AML patients with 11q23/KMT2A (MLL)-rearrangement(KMT2A-r AML). Patients who had received prior cytotoxic therapy, or carried a prior diagnosis of any myeloid neoplasm were excluded from this study. The study was approved by the local Ethics Committee and conducted in accordance with the Declaration of Helsinki.
Cytogenetics, fluorescence in situ hybridization analysis and PCR
Cytogenetic analyses using standard G or R-banding techniques were performed on bone marrow (BM) samples as part of the diagnostic work-up. Karyotyping results were described according to the International System for Human Cytogenetic Nomenclature (ISCN) 2005 [Citation19]. Fluorescence in situ hybridization (FISH) analysis was performed using dual color, break apart probes for detection of rearrangements of KMT2A (MLL), and dual color, dual fusion probe set for detection of t(8;21)/RUNX1T1-RUNX1 fusion, following standard laboratory procedures. A total of 200 cells were counted. 41 fusion genes were detected by multiple reverse transcription PCR amplification, which was performed as 8 parallel multiplex reactions on 7500 Real-time PCR System (Applied Biosystems), as previous report [Citation20].
Gene mutation analysis
Genomic DNA was extracted from fresh BM or peripheral blood (PB) samples using the TIANamp Blood DNA Kit (TIANGEN, Cat#DP318), according to the manufacturer’s instructions. The purity (2.0 > OD260/280 > 1.8) and yield (50 ng per sample) of genomic DNA met the sequencing requirements. A sensitive next-generation amplicon deep-sequencing assay was used with an Illumina next-generation sequencer. A high depth of coverage (1000×) was obtained for 112 genes, including the whole coding regions that are related to hematological malignancies involved in the following functional categories: signaling pathways, epigenetic regulators, transcription factors, spliceosomes, cohesin complex, tumor suppressors, and NPM1. A variant allele frequency (VAF) >3% was used as the threshold for calling single nucleotide variants. Altered DNA sequences were deemed mutations or variants in IGV software analysis and were identified by the COSMIC database, excluding dbSNPs. CEBPA, NPM1, and FLT3-ITD mutations were identified by polymerase chain reaction (PCR) followed by direct Sanger sequencing to avoid the possible false-negative results due to the limitation of bioinformatics analyses of NGS, as previously described [Citation21,Citation22].
Treatment
A total of 35 de novo AML patients with sole trisomy 8 were included in the study. 20 cases received standard chemotherapy regimen (DA/IA, daunorubicin/idarubicin 60/10 mg/m2 d1-3, cytarabine 100 mg/m2 d1-7) for induction therapy, one patient was prescribed with reduced intensity DA/IA (daunorubicin/idarubicin 45/6–8 mg/m2 d1-3, cytarabine 100 mg/m2 d1-7) regimen, three patients received HAD regimen (Homoharringtonine 2 mg/m2 d1-7, cytarabine 100 mg/m2 d1-7, daunorubicin 40 mg/m2 d1-3), 11 patients received decitabine-based induction due to older age or underlying comorbidity.
Reponse and follow up
Complete remission (CR) was defined according to standard criteria as less than 5% blasts in bone marrow aspirates with evidence of maturation of cell lines and restoration of peripheral blood counts, a peripheral blood platelet count >100 × 109/L, an absolute neutrophil count >1.0 × 109/L, and the absence of extramedullary disease [Citation23]. Early mortality was defined as death within 30 days after induction before. Relapse was defined as ≥5% blasts in the BM or the development of extramedullary leukemia. Disease-free survival (DFS) was defined as the time from the date of CR to relapse or death of any cause or the last follow-up. Overall survival (OS) was defined as the time from the start date of induction therapy to death from any cause or last follow-up.
Statistical analysis
Statistical significance was analyzed using Student’s t-test to compare differences of the continuous variables in normal distribution. Mann–Whitney test was used for the comparison of continuous variables in abnormal distribution. Fisher exact test (less than 5 cases per group) or chi-square test were used to calculate statistical significance for categorical variables. The distributions of OS and DFS were plotted by the Kaplan–Meier (K–M) method, and the log-rank test was applied to estimate differences between groups. Univariate and multivariate analyses of survival were carried out by employing the Cox regression model. GraphPad Prism 8.0 (GraphPad Software, San Diego, CA) and SPSS (version 25.0) software (IBM Corporation, Armonk, NY, USA) were used for statistical analysis. Factors that fulfilled the prespecified assumption with P-values <0.1 from univariate analyses were further included as covariates in multivariate Cox regression models. All P values were calculated with the use of two-sided tests and P < 0.05 were considered significant.
Results
Patient characteristics
A total of 35 de novo AML patients (2.9%) with trisomy 8 sole aberration were identified. Of the patients, 18 were male and 17 were female. The median age was 50 years (range: 18–71 years), with 26 patients being < 60 years and 9 patients being ≥ 60 years. According to the French–American–British (FAB) classification, 7, 7, 3, 12, and 6 patients were diagnosed with M1, M2, M4, M5, and unclassified types, respectively. 34.3% of sole +8 AML patients presented with the FAB-M5 subtype, which occurred at a distinctively higher rate than that in NK patients (34.3% vs. 20.6%, P = 0.137). AML patients with isolated trisomy 8 showed lower hemoglobin levels than NK AML patients(P = 0.001), and advanced age than AML patients with t(8;21), or KMT2A-rAML patients(P = 0.001, 0.009). Platelet counts were higher in the sole +8 AML than that in AML with t(8;21) (median: 46.0 × 109/L vs. 26.0×109/L, P = 0.0002), but not in NK AML and KMT2A-r AML(P = 0.440, 0.278). No significant differences were identified regarding sex, white blood cell (WBC) count between the sole +8 AML group and other AML groups. and lists different categories of clinical information of the enrolled patients.
Table 1. Clinical characteristics of AML patients with trisomy 8 as the sole chromosome abnormality.
Table 2. Pretreatment clinical characteristics of AML patients with isolated +8 and comparison by different karyotype groups.
Spectrum of mutations
Based on NGS, a total of 97.14% (34/35) of the sole +8 AML patients harbored at least one mutation, which reached an average number of 3.8(range, 0–10) per patient: two patients (5.7%) had 1 mutation, seven patients (20%) had 2 mutations, seven patients (20%) had 3 mutations, and eighteen patients (51.4%) had 4 or more mutations. The most frequently mutated genes were DNMT3A(37.1%), RUNX1(28.6%), FLT3-ITD(28.6%), IDH2(22.9%), NPM1(17.1%), ASXL1 (14.3%), CEBPA(14.3%,), IDH1(11.4%), and TET2(11.4%). Other genes had a mutation prevalence of < 10%. Functionally, mutations involved in epigenetic regulators (54.3%), transcription factors (51.4%), and signaling pathways (40%) were found frequently in the sole +8 AML patients.
As listed in , the sole +8 AML patients exhibited more mutations in RUNX1 (28.6% vs. 4.8%, P = 0.001) and ASXL1 (14.3% vs. 4.8%, P = 0.039) by comparing with NK AML patients. Epigenetic regulator mutations in IDH2 and DNMT3A were significantly more frequent in the sole +8 AML patients than in AML with t(8;21) and KMT2A-r AML(all P < 0.05), but similar to NK AML patients. In contrast, both KIT and NRAS mutations were less common in the sole +8 AML as compared to AML with t(8;21)(P = 0.001 and 0.044). KRAS mutation was detected only in one patient, the frequency was significantly lower than that in KMT2A-r AML(2.9% vs. 26.8%, P = 0.023).
Table 3. Molecular characteristics of AML patients with isolated +8 and comparison by different karyotype groups.
The relationship between clinical features and mutations
As listed in , the sole +8 AML patients with RUNX1 and IDH2 mutations showed significantly lower WBC counts (P = 0.0203 and 0.039, respectively) as compared to the corresponding wild-type groups, while FLT3-ITD mutations was significantly associated with higher WBC counts (46.0 vs. 5.5 × 109/L, P = 0.00001). Patients harboring TET2 mutations showed a male preponderance of 3:1, but IDH1 mutations were associated with female preponderance (P = 0.015). No significant differences were identified regarding sex, age, hemoglobin, WBC and platelet counts between the DNMT3A- or ASXL1-mutated groups and corresponding wild-type groups.
Table 4. Clinical characteristics of the common mutations in the sole +8 AML patients.
Response to induction therapy and survival analysis
Of 35 AML patients with isolated +8, 45.7% patients achieved CR after the first induction therapy. The CR rate of patients with FLT3-ITD or IDH1 mutation was significant lower than that in the corresponding wild-type cases (P = 0.047, 0.005), while patients harboring TET2 mutations showed much higher CR rate than the wild-type cases(P = 0.00011). No differences were observed in the CR rate between patients with or without DBMT3A, RUNX1, IDH2, and ASXL1 mutations.
The prognosis of the sole +8 AML patients was assessed in terms of OS and DFS. Of 35 patients with isolated +8, the complete follow-up data was available in 30 patients. The median follow-up period was 16.0 months (1.0–52.0 months). The median OS and DFS were 18.0 (95% CI: 10.8–25.2) and 10 (95% CI: 6.7–13.3) months, respectively. According to K–M curves, a significant difference in DFS was only observed for allo-HSCT (7 months vs. NR, P = 0.001), but not for age, WBC count, and gene mutation. Patients who received allo-HSCT had a significantly better OS and DFS than patients who did not undergo allo-HSCT (NR vs. 12 months, P < 0.001; NR vs.7 months, P = 0.001). OS and DFS were also compared in patients with or without specific gene mutations. Only FLT3-ITD mutation was significantly associated with inferior OS (9 vs. 26 months, P = 0.047). There were no significant differences in OS according to the gene mutation status of DNMT3A (16 vs. 18 months, P = 0.843), RUNX1 (14 vs. 20 months, P = 0.432), IDH2 (16 vs. 18 months, P = 0.409), and ASXL1 (16 vs. 18 months, P = 0.458). In multivariable analysis, FLT3-ITD mutations and allo-HSCT were independent prognostic markers for OS ( and ).
Figure 1. Landscape of mutated genes in acute myeloid Leukemia with trisomy 8 as the sole chromosome abnormality. (A) Distribution of acquired mutations into functional categories.(B)Frequency of gene mutations in sole +8 AML and NK AML. Each bar represents a distinct mutated gene. (C) Spectrum of mutated genes in sole +8 AML divided into different mutational categories; each column represents one sample sequenced here.
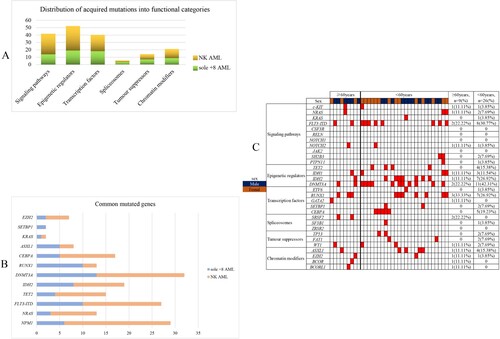
Figure 2. Kaplan–Meier estimates of overall survival (OS) according to gene status, age, WBC and treatment (allo-HSCT or not).
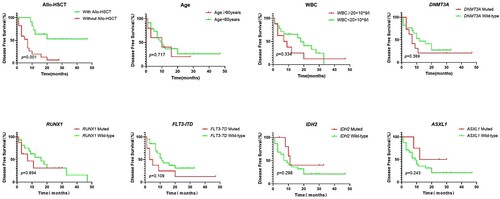
Figure 3. Kaplan–Meier estimates of disease-free survival (DFS) according to gene status, age, WBC and treatment (allo-HSCT or not).
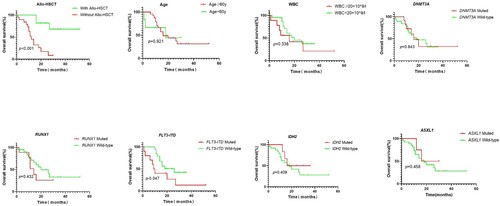
Table 5. Univariate and multivariate COX analysis of factors predicting for OS and DFS.
Discussion
In the present study, we firstly observed that the sole +8 AML showed lower hemoglobin levels compared to other AML patients, which are different from Farag SS et al. and Wolman SR et al. [Citation16,Citation24]. A recent study concluded that pediatric AML trisomy 8 was associated with monocytic differentiation of the AML cells (FAB M5) [Citation25], we also found that the sole +8 AML patients were more likely to present as AML-M5 compared to NK AML. Further analysis of larger collections of sole trisomy 8 AML cases will be necessary to validate our observations.
Becker H et al. demonstrated that 94% of the sole +8 AML patients harbored at least one mutation, the most frequently mutated genes were RUNX1 (32%), ASXL1 (29%), FLT3 (specifically FLT3-ITD: 29%), IDH2 (26%), DNMT3A (25%), and NPM1 (22.5%) [Citation17]. Zong XP et al. demonstrated that the frequency of ASXL1 mutation was identified in 19.2% (15/78) of AML patients with trisomy 8 as the sole aberration or accompanied with other anomalies by sequencing analysis [Citation26]. RUNX1 mutation seem to occur quite frequent in AML with trisomy 8, reporting an incidence varying from 25% to 35% [Citation4,Citation11,Citation27–29]. Alpermann T et al. showed that +8 sole patients harbored more often ASXL1 mutations (46.8% vs. 11.7%, P < 0.001) and RUNX1 mutations (27.8% vs. 15.1%, P = 0.009), but less frequent FLT3-ITD (19.0% vs. 33.3%, P = 0.038), NPM1 mutations (17.7% vs.55.4%, P < 0.001), and double-mutated CEBPA (0 vs. 5.3%, P = 0.038) than NK patients [Citation30]. Our data showed that the incidence of RUNX1 mutation was consistent with data reported in Becker H et al. or Tamara A et al. while the ASXL1 mutations were less frequent in our study than that in study of Becker H et al. or Tamara A et al. We also found that ASXL1 and RUNX1 mutations were more common in the sole +8 AML than that in NK AML. However, no co-mutation was as tightly associated with +8 AML as reported for AML with other numeric aberrations, for example, +11 and MLL-PTD [Citation31], +13 and RUNX1 mutations [Citation32]. Future studies may determine whether +8 favors acquisition of RUNX1 and ASXL1 mutations or whether NK AML with such mutations is prone to the gain of +8.
In AML, genes involved in DNA methylation such as DNMT3A, TET2, IDH1, and IDH2 are frequently mutated [Citation33,Citation34]. Study from Caramazza D et al. suggested a possible association between IDH mutations and trisomy 8 in AML and MDS but not in MPN or MDS/MPN; IDH mutational frequencies in trisomy-8-associated AML and MDS (13–27%) were substantially higher than those of trisomy-8-associated MPN (3%) or MDS/MPN (0%) and also a control group with AML or MDS without isolated trisomy 8 (0–7%) [Citation35]. In our study, the mutation frequencies of DNMT3A, IDH1, IDH2 were significantly higher than those of AML with t(8;21) or KMT2A-r AML, but not NK AML. Further investigations are needed to reveal the relation between trisomy 8 and mutations in epigenetic regulators.
The remission rates for trisomy 8 AML varied widely among studies ranging from 23% to 91% [Citation15,Citation24,Citation36]. Median overall survival time for all patients with +8 was 8.3–25 months [Citation4,Citation15,Citation24,Citation36]. We showed that a lower CR rate of 45.71% and median OS of 18 months, these findings which were consistent with data from Jaff N et al. [Citation36] and Lazarevic VL et al. [Citation4]. It is currently unknown whether molecular markers allow risk stratification in sole +8 AML, and only limited prognostic data on +8 as a sole abnormality are available. Becker H et al. showed that FLT3-ITD status was the only variable associated with DFS; patients with FLT3-ITD had worse DFS than those without (P = 0.054). Both FLT3-ITD and TET2 mutations were associated with worse OS. Among older patients, TET2 mutation status was the only significant marker for CR and DFS [Citation17]. Zong XP et al. observed that ASXL1 mutations are associated with poor prognosis in de novo AML with trisomy 8 as the sole aberration or accompanied with one anomaly which did not include the favorable cytogenetic abnormalities [Citation26]. We also tested the prognostic significance of the various markers in univariable and multivariable models. In our series, The TET2 mutations predicted a higher CR rate, a result contrary to study from Becker H et al. [Citation17]. We found that FLT3-ITD mutation was significantly associated with inferior OS, however, there were no significant differences in OS according to the gene mutation status of RUNX1, DNMT3A, ASXL1, IDH2, and TET2. Thus, AML patients with FLT3-ITD and co-occurring trisomy 8 seem to have a special inferior outcome. The reason for the inconsistency might be due to the age distribution of the cohort, the patient enrollment criteria, the treatment regimen, the small sample size, the regions, the research background.
Data from Schaich M et al. demonstrated that allo-HSCT was independent prognostic factor for longer leukemia-free survival (LFS) compared to the patients who underwent autologous transplantation and patients who were treated with chemotherapy only [Citation37]. Our results also showed that allo-HSCT could improve the OS and DFS, and was an independent factor for better OS. The finding might be useful for the guidance of treatment decisions.
In summary, isolated trisomy 8 is relatively rare in de novo AML. We examined the mutational landscape of the sole +8 AML patients, showing some differences in genetic profiles between these subgroups and NK AML, AML with t(8;21), and KMT2A-r AML, identifying molecular abnormalities that predict OS, and determining independent prognostic factors. However, due to the small cohorts studied, further studies are warranted to clarify these findings.
Author contributions
HongYing Chao and XuZhang Lu conceived the study and designed experimental procedures. Zheng Wang and ZhuXia Jia collected data. Jie Liu and WenMin Han analyzed data and wrote the paper. HongJie Shen and Liujun Cao performed statistical analysis. HaiYing Hua and XiaoHui Cai revised and edited the manuscript. All authors reviewed and approved the final version.
Statement of ethics
Our analysis was performed in accordance with the World Medical Association Declaration of Helsiniki and Ethics Committee approval of Changzhou No.2 People’s Hospital was obtained ([2015]KY004-01). Patients or their lineal relatives involved in this study have given their written informed consent.
Supplemental Material
Download MS Word (80 KB)Acknowledgements
The authors thank all the physicians who provided leukemia samples and clinical data.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
All data generated or analysed during this study are included in this article or its supplementary material files. Further enquiries can be directed to the corresponding author.
Additional information
Funding
References
- Döhner H, Weisdorf DJ, Bloomfield CD. Acute myeloid leukemia. N Engl J Med. 2015;373(12):1136–1152.
- Ley TJ, Miller C, Ding L, et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med. 2013 May;368(22):2059–2074.
- Grimwade D, Hills RK, Moorman AV, et al. Refinement of cytogenetic classification in acute myeloid leukemia: determination of prognostic significance of rare recurring chromosomal abnormalities among 5876 younger adult patients treated in the United Kingdom medical research council trials. Blood. 2010 July;116(3):354–365.
- Lazarevic VL, Rosso A, Juliusson G, et al. Incidence and prognostic significance of isolated trisomies in adult acute myeloid leukemia: a population-based study from the Swedish AML registry. Eur J Haematol. 2017 Jan;98(5):493.
- Xiao Y, Wei J, Chen Y, et al. Trisomy 8 is the most frequent cytogenetic abnormality in de novo myelodysplastic syndrome in China. Onkologie. 2012 Feb;35(3):100–106.
- Yan XF, Wang L, Jiang LX, et al. Clinical significance of cytogenetic and molecular genetic abnormalities in 634 Chinese patients with myelodysplastic syndromes. Cancer Med. 2021 Feb;10(5):1757–1771.
- Such E, Cervera J, Costa D, et al. Cytogenetic risk stratification in chronic myelomonocytic leukemia. Haematologica. 2011 Feb;96:375–383.
- Wang W, Cortes JE, Lin P, et al. Impact of trisomy 8 on treatment response and survival of patients with chronic myelogenous leukemia in the era of tyrosine kinase inhibitors. Leukemia. 2015 Nov;29(11):2263–2266.
- Grinfeld J, Nangalia J, Baxter EJ, et al. Classification and personalized prognosis in myeloproliferative neoplasms. N Engl J Med. 2018 Oct;379(15):1416–1430.
- Mrózek K, Heerema NA, Bloomfifield CD. Cytogenetics in acute leukemia. Blood Rev. 2004 May;18(2):115–136.
- Hemsing AL, Hovland R, Tsykunova G, et al. Trisomy 8 in acute myeloid leukemia. Expert Rev Hematol. 2019 Aug;12(11):947–958.
- Yin CC, Cortes J, Barkoh B, et al. T(3;21)(q26;q22) in myeloid leukemia: An aggressive syndrome of blast transformation associated with hydroxyurea or antimetabolite therapy. Cancer. 2006 Mar;106(8):1730–1738.
- Noort S, Zimmermann M, Reinhardt D, et al. Prognostic impact of t(16;21)(p11;q22) and t(16;21)(q24;q22) in pediatric AML: a retrospective study by the I-BFM study group. Blood. 2018 Oct;132(15):1584–1592.
- Coenen EA, Raimondi S, Harbott J, et al. Prognostic significance of additional cytogenetic aberrations in 733 de novo pediatric 11q23/MLL-rearranged AML patients: results of an international study. Blood. 2011 Sep;117(26):7102–7111.
- Elliott MA, Letendre L, Hanson CA, et al. The prognostic significance of trisomy 8 in patients with acute myeloid leukemia. Leuk Lymphoma. 2002 Mar;43(3):583–586.
- Farag SS, Archer KJ, Mrozek K, et al. Isolated trisomy of chromosomes 8, 11, 13 and 21 is an adverse prognostic factor in adults with de novo acute myeloid leukemia: results from cancer and leukemia group B 8461. Int J Oncol. 2002 Nov;21(5):1041–1051.
- Becker H, Maharry K, Mrózek K, et al. Prognostic gene mutations and distinct gene- and microRNA-expression signatures in acute myeloid leukemia with a sole trisomy 8. Leukemia. 2014 Aug;28(8):1754–1758.
- Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016 May;127(20):2391–2405.
- Mccarthy C.. ISCN 2005: An International System for Human Cytogenetic Nomenclature [Book Review]. Australian J Med Sci. 2006;27(4):153.
- Wang K, Zhou F, Cai X, et al. Mutational landscape of patients with acute myeloid leukemia or myelodysplastic syndromes in the context of RUNX1 mutation. Hematology. 2020 May;25(1):211–218.
- Lin LI, Chen CY, Lin DT, et al. Characterization of CEBPA mutations in acute myeloid leukemia: most patients with CEBPA mutations have biallelic mutations and show a distinct immunophenotype of the leukemic cells. Clin Cancer Res. 2005 Feb;11(4):1372–1379.
- Chen W, Konoplev S, Medeiros LJ, et al. Cuplike nuclei (prominent nuclear invaginations) in acute myeloid leukemia are highly associated with FLT3 internal tandem duplication and NPM1 mutation. Cancer. 2009 Aug;115(23):5481–5489.
- O’Donnell MR, Tallman MS, Abboud CN, et al. Acute myeloid leukemia, version 3.2017, NCCN clinical practice guidelines in oncology. J Natl Compr Canc Netw. 2017 Jan;15:926–957.
- Wolman SR, Gundacher H, Appelbaum FR, et al. Impact of trisomy 8 (+8) on clinical presentation, treatment response, and survival in acute myeloid leukemia: a Southwest oncology group study. Blood. 2002 Feb;100(1):29–35.
- Laursen ACL, Sandahl JD, Kjeldsen E, et al. Trisomy 8 in pediatric acute myeloid leukemia: A NOPHO-AML study. Genes Chromosomes Cancer. 2016 Apr;55(9):719–726.
- Zong XP, Yao H, Wen LJ, et al. ASXL1 mutations are frequent in de novo AML with trisomy 8 and confer an unfavorable prognosis. Leuk Lymphoma. 2017 Jan;58(1):204–206.
- Schnittger S, Dicker F, Kern W, et al. RUNX1 mutations are frequent in de novo AML with noncomplex karyotype and confer an unfavorable prognosis. Blood. 2011 Aug;117(8):2348–2357.
- Tang JL, Hou HA, Chen CY, et al. AML1/RUNX1 mutations in 470 adult patients with de novo acute myeloid leukemia: prognostic implication and interaction with other gene alterations. Blood. 2009 Oct;114(26):5352–5361.
- Stengel A, Kern W, Meggendorfer M, et al. Number of RUNX1 mutations, wild-type allele loss and additional mutations impact on prognosis in adult RUNX1-mutated AML. Leukemia. 2018 Feb;32(2):295–302.
- Alpermann T, Haferlach C, Eder C, et al. AML with gain of chromosome 8 as the sole chromosomal abnormality (+8sole) is associated with a specific molecular mutation pattern including ASXL1 mutations in 46.8% of the patients. Leuk Res. 2015 Mar;39(3):265–272.
- Caligiuri MA, Strout MP, Schichman SA, et al. Partial tandem duplication of ALL1 as a recurrent molecular defect in acute myeloid leukemia with trisomy 11. Cancer Res. 1996 Mar;56(6):1418–1425.
- Dicker F, Haferlach C, Kern W, et al. Trisomy 13 is strongly associated with AML1/RUNX1 mutations and increased FLT3 expression in acute myeloid leukemia. Blood. 2007 May;110(4):1308–1316.
- Im AP, Sehgal AR, Carroll MP, et al. DNMT3A and IDH mutations in acute myeloid leukemia and other myeloid malignancies: associations with prognosis and potential treatment strategies. Leukemia. 2014 Apr;28(9):1774–1783.
- Dunlap JB, Leonard J, Rosenberg M, et al. The combination of NPM1, DNMT3A and IDH1/2 mutations leads to inferior overall survival in AML. Am J Hematol. 2019 May;94(8):913–920.
- Caramazza D, Lasho TL, Finke CM, et al. IDH mutations and trisomy 8 in myelodysplastic syndromes and acute myeloid leukemia. Leukemia. 2010 Sep;24(12):2120–2122.
- Jaff N, Chelghoum Y, Elhamri M, et al. Trisomy 8 as sole anomaly or with other clonal aberrations in acute myeloid leukemia: impact on clinical presentation and outcome. Leuk Res. 2007 Jan;31(1):67–73.
- Schaich M, Schlenk RF, Al-Ali HK, et al. Prognosis of acute myeloid leukemia patients up to 60 years of age exhibiting trisomy 8 within a non-complex karyotype: individual patient data-based meta-analysis of the German acute myeloid leukemia intergroup. Haematologica. 2007 May;92(6):763–770.