
ABSTRACT
Objectives
Atypical hemolytic uremic syndrome (aHUS) is characterized by a triad of thrombocytopenia, microangiopathic hemolytic anemia, and acute renal failure resulting from platelet thrombi in the microcirculation of the kidney and other organs, in the absence of a preceding diarrheal illness. This report describes a case in which copy number variation (CNV) analysis using next-generation sequencing (NGS) identified the CFHR3/CFHR1 deletion in a patient with aHUS.
Methods
A 49-year-old Korean female was diagnosed with aHUS based on clinical findings, including schistocytes in peripheral blood and marked thrombocytopenia, suggesting the presence of thrombotic microangiopathy, elevated serum lactate dehydrogenase, and acute kidney injury. Sequence variants and CNV generated from NGS data were estimated to determine if there was a potential genetic cause. Multiplex ligation-dependent probe amplification (MLPA) was conducted to confirm the CFHR3/CFHR1 deletion identified by NGS with CNV analysis.
Results
No known or novel pathogenic single nucleotide variant or small insertion/deletion that would be predicted to have damaging effects that could lead to aHUS were identified. However, CNV analysis of NGS data identified the heterozygous CFHR3/CFHR1 deletion. MLPA confirmed this loss of one copy number between the CFHR3 and the CFHR1 genes on chromosome 1q31.3.
Conclusion
We genetically diagnosed a Korean woman harboring a heterozygous CFHR3/CFHR1 deletion of a known causative gene for aHUS. Our report emphasizes the need for CNV analysis of NGS data and gene dosage assays, such as MLPA, to evaluate large-scale deletions or duplications and generate hybrid CFH genes in patients with suspected aHUS.
Introduction
Atypical hemolytic uremic syndrome (aHUS) is characterized by a triad of thrombocytopenia, microangiopathic hemolytic anemia, and acute renal failure that results from platelet thrombi in the microcirculation of the kidney and other organs, in the absence of a preceding diarrheal illness [Citation1]. Several large cohort studies in different ethnic populations have reported complement gene mutations in up to 60% of aHUS patients [Citation2,Citation3]. The most commonly mutated gene in aHUS is CFH, which encodes the complement regulator of complement factor H–related proteins (CFHR1 to 5) residing in a centromeric 355 kb segment on chromosome 1q32. Disease-causing mutations are detected in up to 40% of familial and 25% of sporadic cases [Citation2,Citation3]. It is likely that mutation of CFH, CFI, CFB, C3, CD46, or THBD confers a predisposition to developing aHUS rather than directly leading to the disease. Particularly, a hybrid CFH/CFHR3 gene resulting from a microhomology-mediated deletion in familial aHUS has been reported [Citation4]. Molecular analysis of this region provides evidence of multiple independent large genomic duplications, also known as low-copy repeats, resulting in a high degree of sequence identity between CFH and CFHR1 to CFHR5 [Citation5]. In addition, non-enteric bacterial (Streptococcus pneumoniae accounts for 40% of aHUS) and viral infections, malignancies (∼6%), drugs, pregnancy, and transplantation that trigger complement activation can precipitate an acute occurrence in those with predisposing genetic evidence [Citation6]. In patients in whom the pathogenic mutation(s) related to aHUS has been identified, monitoring of laboratory findings such as hemoglobin, platelet count, and serum concentrations of C3, C4, LDH, haptoglobin, and creatinine is required after exposure to potential triggering events [Citation7]. Eculizumab, a first-in-class humanized monoclonal anti-C5 antibody is recommended to treat aHUS and to induce remission of aHUS refractory to plasma manipulation [Citation8]. While plasma exchange or infusion can decrease mortality, plasma dependence or resistance also can occur [Citation9].
Deletions within genes, occurring through both microhomology-mediated end joining and non-allelic homologous recombination, result in the formation of hybrid genes (CFH/CFHR1, CFHR1/CFH, CFH/CFHR3, CFHR3/CFHR1) related to aHUS [Citation10]. Thus, NGS panel data can be used as a CNV screening step in a genetic diagnostics setting, and this screening step has the potential to improve the genetic diagnosis of aHUS, even though most CNV tools were designed to work with whole exome or whole genome data and struggle with the sparser data from NGS panels used in routine genetic testing [Citation11]. This report describes a case in which copy number variation (CNV) analysis using next-generation sequencing (NGS) identified the CFHR3/CFHR1 deletion in a patient with aHUS.
Case presentation
A 49-year-old, previously healthy Korean woman with no remarkable medical history except for being born with a single kidney was admitted to the emergency room with generalized weakness that had persisted for one month. She had experienced weight loss of 5–6 kg, petechiae on the abdomen, and progressive edema on both legs over the last month. Over the last week, the patient had been unable to complete normal life activities due to a rapidly progressing generalized weakness and fever. Although the patient had a decreased performance status, no signs or symptoms indicated neurologic deficits. Laboratory tests revealed marked pancytopenia, reticulocytosis, elevated lactate dehydrogenase level, and kidney injury. The direct and indirect Coombs’ tests were negative, and an immunological profile was within normal ranges except for hypocomplementemia (C3, 62.6 mg/dL) and positive lupus anticoagulant (). Unfortunately, anti-CFH autoantibody testing was not available. Examination of peripheral blood smear revealed numerous schistocytes (fragmented red blood cells) and a markedly decreased number of platelets, suggesting the presence of thrombotic microangiopathy (TMA). The patient did not have a history of bloody diarrhea, and the stool assay for Shiga toxin-producing E. coli was negative. The ADAMTS13 activity was within the normal range (54.9%). Based on these findings, a clinical diagnosis of aHUS was made. Immediately after admission, the patient was treated with therapeutic plasma exchange and concomitant corticosteroid therapy (1 mg/kg of prednisone per day). Plasma exchange was continued daily until 10 days after the event, but the resolution of renal dysfunction and normalization of thrombocytopenia were not obtained. Adjunctive therapy with rituximab (375 mg/m2 once weekly for four consecutive weeks) was added and continued for 13 days, in addition to the daily plasma exchange and prednisone treatment. We also tried to obtain approval for the use of eculizumab from the National Health Insurance Corporation (NHIC) of the Korean government, but the approval was delayed and repeatedly rejected due to problematic consistency in fulfilling all criteria for approval. Unfortunately, even after lengthy efforts to access eculizumab for use and extensive supportive care, the patient’s status progressively deteriorated, and she died 34 days after admission because of multi-organ failure. Because approximately 60% of aHUS cases result from a genetic origin [Citation7], clinical screening of the patient’s family for hereditary aHUS was necessary. However, other family members presented with no clinical symptoms and declined testing, thus genetic counseling and segregation analysis were not evaluated.
Table 1. Results of laboratory blood analyses in a Korean patient with atypical hemolytic uremic syndrome.
Methods
Next-generation gene panel sequencing
This study was approved by the Institutional Review Board (IRB) of Jeonbuk National University Hospital (IRB No. 2021-01-052). The requirement for written informed consent was waived by the IRB because of the anonymous and retrospective nature of this study. To determine the potential genetic cause of the aHUS in our patient, her exomic DNA was analyzed by next-generation gene panel sequencing that consisted of the CFH, C3, CFB, CFI, ADAMTS13, CD46, CFHR1, CFHR2, CFHR3, CFHR5, DGKE, MMACHC, MMADHC, MMUT, PIGA, PLG, and THBD genes responsible for aHUS. Paired-end sequencing was conducted on the Illumina HiSeq2500 (Illumina, San Diego, CA, USA) to detect the variant, given the suspicion of aHUS at the Green Cross Genome (Yongin, Korea). Base calling, alignment, variant calling, annotation, and quality control reporting were performed using the Genome Analysis Tool Kit best-practice pipeline workflow for germline short variant discovery (https://gatk.broadinstitute.org/hc/en-us). Interpretation of sequence variants was manually reviewed by medical laboratory geneticists according to standards and guidelines from the Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology [Citation12]. Historically, aHUS has been associated with a chromosomal deletion of a large genomic fragment (up to 84 kb) in the regulators of complement activation gene clusters at chromosome 1q32 [Citation13,Citation14]. Thus, CNV analysis from panel-based NGS data was estimated using ExomeDepth, call CNVs from targeted sequence data [Citation15], and VisCap, inference, and visualization of germline CNVs from targeted clinical sequencing data [Citation16].
Multiplex ligation-dependent probe amplification
Multiplex ligation-dependent probe amplification (MLPA) was performed to confirm the CFHR3/CFHR1 deletion identified by NGS with CNV analysis using a SALSA® MLPA® Probemix P236-A3 ARMD mix-1 (MRC-Holland, Amsterdam, The Netherlands) according to the manufacturer’s protocol. Capillary electrophoresis and fragment analysis were conducted on the 3730XL Genetic Analyzer (Applied Biosystems, Carlsbad, CA, USA). The electropherogram data were analyzed using GeneMarker software ver. 3.0.1 (Softgenetics, State College, PA, USA) according to the manufacturer’s instructions. The resulting peak intensities were normalized to those of the sex-matched normal DNA and manufacturer control probes as a reference. A probe-to-peak ratio between 0.8 and 1.2 was defined as a normal copy number (wild-type), and a ratio between 0.4 and 0.65 represented a heterozygous deletion (loss of one copy number).
Results
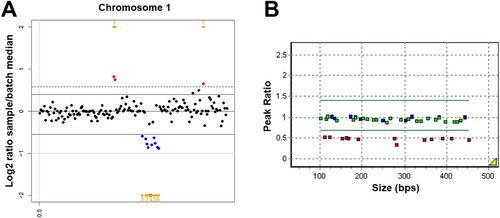
A yield on target of 50,987,990 reads was generated from the patient’s sample by estimating the sequence quality along with all sequences. The mean read length was 158 bp, and the percentage of bases above 30x was 98.2%. No known or novel pathogenic single nucleotide variant or small insertion/deletion predicted to have a damaging effect that could lead to aHUS were identified. However, both the two CNV detection tools identified a heterozygous partial deletion of chromosome 1q31.3, including a region from the CFHR3 and the CFHR1 genes in the patient, with a Z ratio between −0.5 and −1 ((A)). MLPA was additionally performed to confirm this CFHR3/CFHR1 deletion and demonstrated a heterozygous deletion between the CFHR3 (NM_021023.5; 5UTR, exon 1, exon 2, exon 3, intron 4, and exon 6) and the CFHR1 (NM_002113.2; intron1, intron 3, exon5, and exon 6) genes on chromosome 1q31.3 in the patient ((B)). As a result, our case was diagnosed as hereditary aHUS caused by CFHR3/CFHR1 deletion.
Figure 1. The heterozygous CFHR3/CFHR1 deletion identified by copy number variation (CNV) analysis using next-generation sequencing and multiplex ligation-dependent probe amplification (MLPA) in a patient with atypical hemolytic uremic syndrome. (A) CNV detection tool identifies a heterozygous partial deletion of chromosome 1q31.3, including a region from the CFHR3 and the CFHR1 genes, with a Z ratio between −0.5 and −1. (B) MLPA demonstrates a heterozygous deletion between the CFHR3 (NM_021023.5; 5UTR, exon 1, exon 2, exon 3, intron 4, and exon 6) and the CFHR1 (NM_002113.2; intron1, intron 3, exon5, and exon 6) genes on chromosome 1q31.3 in the patient.
Discussion
CFHR–Factor H gene cluster variations are associated with several kidney disorders, including aHUS, IgA nephropathy, and C3 glomerulopathy. These associations show the critical roles of the FHR proteins in maintaining glomerular integrity. FHR proteins are complement modulators and complement activators, and FHR1 regulates inflammasome activity [Citation17]. Our patient experienced an acute episode of aHUS caused by heterozygous CFHR3/CFHR1 deletion, without underlying medical conditions that triggered complement activation. Hetero- and homozygous deletions of CFHR3 and CFHR1 through non-allelic homologous recombination events downstream of CFH is related to early-onset aHUS (under 21 years) [Citation13]. The absence of both CFHR3 and CFHR1 proteins has been reported in approximately 10% of aHUS [Citation14]. CFHR3/CFHR1 plasma deficiency is related to issues for most affected patients due to the formation of anti-CFH autoantibodies. These antibodies bind to both CFHR1 and to the CFH C-terminal, reduce CFH binding to C3b, and enhance alternative pathway-dependent lysis of sheep erythrocytes without influencing fluid-phase cofactor activity [Citation14]. However, the causal link between CFHR3/CFHR1 deletion and CFH autoantibodies is not well known.
Patients with hereditary aHUS experience recurrent relapse even after complete recovery from the presenting episode, and about two-thirds of them progress to end-stage renal disease [Citation6]. Thus, complement and complement regulatory gene mutation analyses such as CFH, C3, CFB, CFI, CFHR, CD46, and THBD or assessment of neutralizing autoantibody response to Factor H could be useful for predicting the risk of aHUS [Citation14]. In addition to sequencing a clinically enhanced exome to enable targeted disease-specific variant analysis, the application of CNV detection algorithms using various tools in a routine targeted NGS diagnostic settings can facilitate immediate improvement in clinical care for individuals with heterogeneous genetic diseases [Citation11,Citation15,Citation18]. NGS is increasingly used for clinical evaluation of patients presenting with TMA because it allows simultaneous interrogation of multiple complement and coagulation pathway genes known to be associated with disease [Citation19,Citation20]. Similarly, genetic testing for multiple genes in patients with aHUS that are negative for anti-FH antibodies reveals multiple candidate variations that require prioritization [Citation21].
Although genetic defect of CFH is a common cause of aHUS, acquired functional defects of CFH due to the development of anti-CFH autoantibodies have been reported in 6–11% of aHUS cases [Citation22]. Particularly, plasma from aHUS patients with anti-CFH antibody caused increased platelet aggregation, complement deposition and increased expression of markers for platelet activation on the platelet surface [Citation23]. Furthermore, anti-CFH antibody itself suppressed thrombosis, while other plasma factor, including complement factors could overactivate the platelets, leading to platelet aggregation [Citation24]. Because patients with both homozygous CFHR1/CFHR3 deficiency and anti-CFH autoantibodies may require plasma infusion or plasmapheresis as well as intensive immunosuppressive therapy, anti-CFH autoantibody testing would be useful for screening acquired or hereditary cause, as well as planning a therapeutic strategy in aHUS patients [Citation25].
Eculizumab has been successful for treating paroxysmal nocturnal hemoglobinuria, a disorder of complement-induced hemolytic anemia, which received approval for treating aHUS in the United States and Europe in late 2011 [Citation26]. Eculizumab therapy should be initiated, when the suspected diagnosis of complement-mediated aHUS is not refuted, and patient did not respond to plasma exchange [Citation27], Eculizumab was associated with significant time-dependent improvement in renal function in patients with aHUS [Citation28,Citation29]. Long-term eculizumab treatment is effective in patients with aHUS, with earlier intervention associated with a greater clinical benefit [Citation30]. Moreover, eculizumab therapy can prevent TMA events [Citation31], and prophylactic treatment can prevent post-transplantation aHUS recurrence in those with mutation in circulating factors such as CFB, CFH, CFI, and C3 [Citation32].
Although plasma exchange prophylaxis has been reported to prevent disease recurrence in patients with CFH mutation [Citation33], the effect of this conventional therapy was very limited in our patients. The Korea National Health Insurance Service allows insurance benefits only in patients with aHUS that satisfy the following criteria: (1) TMA (platelet < lower limit of the reference range, schistocytes on peripheral blood smear, hemoglobin <10 g/dL, serum lactate dehydrogenase level >1.5x upper limit of reference range); (2) renal impairment (decreased eGFR > 20% in known chronic kidney disease, serum creatinine > upper limit of reference range based on age and sex in a normal patient); (3) ADAMTS13 activity > 10% before therapeutic plasma exchange, and (4) events were not caused by Shiga toxin-producing E. coli. Unfortunately, our patient did not receive eculizumab therapy because insurance benefit approval was delayed. Thus, the use of eculizumab should be immediately approved if an apparent genetic cause of aHUS is identified, such as the CFHR3/CFHR1 deletion presented in this case.
In conclusion, we genetically diagnosed a Korean woman harboring a heterozygous CFHR3/CFHR1 deletion, which is a known causative gene for aHUS. Our report emphasizes the need to complete CNV analysis of NGS data and gene dosage assays, such as MLPA, to evaluate large-scale deletions or duplications and generate hybrid CFH genes in patients with suspected aHUS.
Acknowledgements
The authors would like to thank Chang-Seok Ki, M.D., Ph.D. and Ju Sun Song, M.D. (Green Cross Genome, Yongin, Korea) for their contributions in technical support for data analysis of next-generation sequencing.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Taylor CM, Chua C, Howie AJ, et al. Clinico-pathological findings in diarrhoea-negative haemolytic uraemic syndrome. Pediatr Nephrol. 2004;19:419–425.
- Maga TK, Nishimura CJ, Weaver AE, et al. Mutations in alternative pathway complement proteins in American patients with atypical hemolytic uremic syndrome. Hum Mutat. 2010;31:E1445–E1460.
- Fremeaux-Bacchi V, Fakhouri F, Garnier A, et al. Genetics and outcome of atypical hemolytic uremic syndrome: a nationwide French series comparing children and adults. Clin J Am Soc Nephrol. 2013;8:554–562.
- Francis NJ, McNicholas B, Awan A, et al. A novel hybrid CFH/CFHR3 gene generated by a microhomology-mediated deletion in familial atypical hemolytic uremic syndrome. Blood. 2012;119:591–601.
- Díaz-Guillén MA, Rodríguez de Córdoba S, Heine-Suñer D. A radiation hybrid map of complement factor H and factor H-related genes. Immunogenetics. 1999;49:549–552.
- Noris M, Caprioli J, Bresin E, et al. Relative role of genetic complement abnormalities in sporadic and familial aHUS and their impact on clinical phenotype. Clin J Am Soc Nephrol. 2010;5:1844–1859.
- Campistol JM, Arias M, Ariceta G, et al. An update for atypical haemolytic uraemic syndrome: diagnosis and treatment. A consensus document. Nefrologia. 2015;35:421–447.
- Fakhouri F, Frémeaux-Bacchi V, Loirat C. Atypical hemolytic uremic syndrome: from the rediscovery of complement to targeted therapy. Eur J Intern Med. 2013;24:492–495.
- Nester CM, Barbour T, de Cordoba SR, et al. Atypical aHUS: state of the art. Mol Immunol. 2015;67:31–42.
- Holmes LV, Strain L, Staniforth SJ, et al. Determining the population frequency of the CFHR3/CFHR1 deletion at 1q32. PLoS One. 2013;8:e60352, 1–7.
- Moreno-Cabrera JM, Del Valle J, Castellanos E, et al. Evaluation of CNV detection tools for NGS panel data in genetic diagnostics. Eur J Hum Genet. 2020;28:1645–1655.
- Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–424.
- Zipfel PF, Edey M, Heinen S, et al. Deletion of complement factor H-related genes CFHR1 and CFHR3 is associated with atypical hemolytic uremic syndrome. PLoS Genet. 2007;3:e41.
- Moore I, Strain L, Pappworth I, et al. Association of factor H autoantibodies with deletions of CFHR1, CFHR3, CFHR4, and with mutations in CFH, CFI, CD46, and C3 in patients with atypical hemolytic uremic syndrome. Blood. 2010;115:379–387.
- Ellingford JM, Campbell C, Barton S, et al. Validation of copy number variation analysis for next-generation sequencing diagnostics. Eur J Hum Genet. 2017;25:719–724.
- Pugh TJ, Amr SS, Bowser MJ, et al. Viscap: inference and visualization of germ-line copy-number variants from targeted clinical sequencing data. Genet Med. 2016;18:712–719.
- Zipfel PF, Wiech T, Stea ED, et al. CFHR gene variations provide insights in the pathogenesis of the kidney diseases atypical hemolytic uremic syndrome and C3 glomerulopathy. J Am Soc Nephrol. 2020;31:241–256.
- Yao R, Yu T, Qing Y, et al. Evaluation of copy number variant detection from panel-based next-generation sequencing data. Mol Genet Genomic Med. 2019;7:e00513.
- Bu F, Borsa NG, Jones MB, et al. High-throughput genetic testing for thrombotic microangiopathies and C3 glomerulopathies. J Am Soc Nephrol. 2016;27:1245–1253.
- Gaut JP, Jain S, Pfeifer JD, et al. Routine use of clinical exome-based next-generation sequencing for evaluation of patients with thrombotic microangiopathies. Mod Pathol. 2017;30:1739–1747.
- Thergaonkar RW, Narang A, Gurjar BS, et al. Targeted exome sequencing in anti-factor H antibody negative HUS reveals multiple variations. Clin Exp Nephrol. 2018;22:653–660.
- Józsi M, Licht C, Strobel S, et al. Factor H autoantibodies in atypical hemolytic uremic syndrome correlate with CFHR1/CFHR3 deficiency. Blood. 2008;111:1512–1514.
- Speth C, Rambach G, Würzner R, et al. Complement and platelets: mutual interference in the immune network. Mol Immunol. 2015;67:108–118.
- Fujisawa M, Yasumoto A, Kato H, et al. The role of anti-complement factor H antibodies in the development of atypical haemolytic uremic syndrome: a possible contribution to abnormality of platelet function. Br J Haematol. 2020;189:182–186.
- Lee BH, Kwak SH, Shin JI, et al. Atypical hemolytic uremic syndrome associated with complement factor H autoantibodies and CFHR1/CFHR3 deficiency. Pediatr Res. 2009;66:336–340.
- Schmidtko J, Peine S, El-Housseini Y, et al. Treatment of atypical hemolytic uremic syndrome and thrombotic microangiopathies: a focus on eculizumab. Am J Kidney Dis. 2013;61:289–299.
- Wijnsma KL, Duineveld C, Wetzels JFM, et al. Eculizumab in atypical hemolytic uremic syndrome: strategies toward restrictive use. Pediatr Nephrol. 2019;34:2261–2277.
- Dedhia P, Govil A, Mogilishetty G, et al. Eculizumab and belatacept for De novo atypical hemolytic uremic syndrome associated with CFHR3-CFHR1 deletion in a kidney transplant recipient: A case report. Transplant Proc. 2017;49:188–192.
- Nozawa A, Ozeki M, Hori T, et al. A heterozygous CFHR3-CFHR1 gene deletion in a pediatric patient With transplant-associated thrombotic microangiopathy Who was treated With eculizumab. J Pediatr Hematol Oncol. 2018;40:e544–e5e6.
- Legendre CM, Licht C, Muus P, et al. Terminal complement inhibitor eculizumab in atypical hemolytic-uremic syndrome. N Engl J Med. 2013;368:2169–2181.
- Fakhouri F, Hourmant M, Campistol JM, et al. Terminal complement inhibitor eculizumab in adult patients with atypical hemolytic uremic syndrome: A single-Arm, open-label trial. Am J Kidney Dis. 2016;68:84–93.
- Krid S, Roumenina LT, Beury D, et al. Renal transplantation under prophylactic eculizumab in atypical hemolytic uremic syndrome with CFH/CFHR1 hybrid protein. Am J Transplant. 2012;12:1938–1944.
- Davin JC, Strain L, Goodship TH. Plasma therapy in atypical haemolytic uremic syndrome: lessons from a family with a factor H mutation. Pediatr Nephrol. 2008;23:1517–1521.