
ABSTRACT
Objectives: Thalassemia, the most common global monogenetic disorder, is highly prevalent in southern China. Epidemiological and molecular characterization of thalassemia is important for designing appropriate prevention strategies in high-risk areas, especially the border area of Guangxi-Yunnan-Guizhou province in southwestern China.
Methods: We recruited 38812 reproductive age couples and screened them for thalassemia. Routine blood tests as well as hemoglobin components and levels were evaluated. In addition, suspected thalassemia were identified by gap polymerase chain reaction (Gap-PCR) and PCR-based reverse dot blot (PCR-RDB).
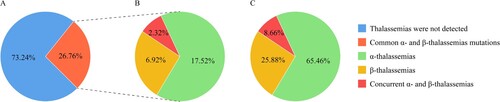
Results: The overall prevalence of thalassemia was 26.76%. Specifically, incidences of α-thalassemia, β-thalassemia, and concurrent α- and β-thalassemia were 17.52%, 6.92%, and 2.32%, respectively. The diagnosed α-thalassemia anomalies were associated with six gene mutations and 25 genotypes. The β-thalassemia anomalies were associated with 12 gene mutations and 15 genotypes. Moreover, among the 1799 concurrent mutated α- and β-thalassemia genes, 95 different genotypes were identified. Couples in which both partners were positive for α-thalassemia and β-thalassemia isotypes were 8.80% and 2.08%, respectively. The proportion of couples at a risk of having children with thalassemia major or intermedia was high.
Conclusions: This study elucidates on the prevalence and molecular characterization of thalassemia in the border area of Guangxi-Yunnan-Guizhou provinces. These findings provide valuable baseline data for genetic counseling and prenatal diagnosis, with the overarching goal of preventing and controlling severe thalassemia.
Introduction
Thalassemia, an autosomal-recessive inherited disorder, is characterized by reduced or absent synthesis of one or several globin peptide chains, which may result in hemolytic anemia [Citation1]. The degree of anemia varies depending on imbalances in production of α- and β-like chains. Thalassemia minor may be asymptomatic or may present mild symptoms, whereas thalassemia intermedia is commonly heterogeneous with symptoms varying between severe transfusion-dependent thalassemia major to the mild thalassemia intermedia form. Babies with thalassemia major either die before birth or are born with serious complications that cause significant psychosocial and economic burden to their families and society [Citation2,Citation3]. Thalassemia major is refractory but preventable, with the only effective interventions involving precision genetic analysis on thalassemia intermediate, and preventing the birth of babies with thalassemia major by detection of carriers and prenatal genetic diagnosis [Citation4,Citation5].
Globally, thalassemia is common in Mediterranean countries, Middle East, Indian subcontinent, and Southeast Asia [Citation6]. In China, thalassemia is prevalent in southern regions, with Guangxi, Yunnan, Guizhou, Guangdong, and Hainan provinces being the most affected [Citation7]. However, currently, there is little information on epidemiology of thalassemia in the border area of Guangxi-Yunnan-Guizhou provinces, an underdeveloped minority nationality region, which greatly affects genetic counseling and prenatal diagnosis. Therefore, we aimed at exploring thalassemia gene mutations, and uncovering the characteristics as well as hematological parameters of thalassemia genotypes in the border area of Guangxi-Yunnan-Guizhou provinces, southwest China, during a 5-year period ranging from 2017 to 2021. We found a high prevalence of thalassemia, with diversity mutation spectrums among people of reproductive age in the border area of Guangxi-Yunnan-Guizhou provinces in southwestern China. Collectively, our findings provide critical insights for genetic counseling, prenatal diagnosis, and prevention of severe thalassemia in this region.
Materials and methods
Ethical statement
This study was reviewed and approved by the Ethical Review Committee of the Affiliated Hospital of YouJiang Medical College for Nationalities (YYFY-LL-2022-001). Prior to being enrolled in the study, signed informed consents were obtained from all participants. All experiments were performed in accordance with the relevant institutional guidelines and regulations.
Study participants
During their antemarital and prenatal examinations, 38812 couples were recruited in this study and screened for thalassemia in the period from January 2017 to December 2021. These participants were from the border area of Guangxi-Yunnan-Guizhou provinces. This border area, which is a multi-ethnic district, includes the Baise city, the northwest region of Guangxi Province, Wenshan Zhuang, and Miao Autonomous Prefecture in southeastern Yunnan Province, as well as Qianxinan Buyi and Miao Autonomous Prefecture in Guizhou Province.
Thalassemia screening
EDTA-K2-anticoagulated peripheral blood samples were collected from each participant. Next, blood cell parameters were determined by full automatic blood cell analysis (XN-1000, Sysmex, Japan), followed by analysis of hemoglobin components and levels by high-performance liquid chromatography (Variant II, BIO-RAD, USA). In conjunction with the local thalassemia prevention and control program, thalassemia screening was positive if it met any one of the following criteria: (1) Corpuscular volume (MCV) <82.5fL; (2) Mean corpuscular Hb (MCH) levels <27pg; (3) Hemoglobin A2 (HbA2) >3.5%; (4) HbA2 < 2.5%; (5) Fetal hemoglobin (HbF) >2.0%; and (6) Abnormal hemoglobin peaks. All individuals who were positive for thalassemia during screening were subjected to genetic testing.
Genetic analysis
Genomic DNA was extracted from peripheral blood using a genomic DNA isolation kit (Yaneng Biological Technology Co., Ltd., Shenzhen, China). The three common types of deletional α-thalassemia (-α3.7, -α4.2, and --SEA) were detected by gap polymerase chain reaction (Gap-PCR). In addition, the three non-deletional types of α-thalassemia [Hb Constant Spring (Hb CS), Hb Quong Sze (Hb QS), and Hb Westmead (Hb WS)] and the 17 β-globin gene mutations [CD41/42 (-TTCT), CD43 (GAG-TAG), IVS-II-654 (C-T), -28 (A-G), -29 (A-G), -32 (C-A), CD71/72 (+A), CD26 (GAG-AAG), CD17 (AAG-TAG), CD31 (-C), CD14/15 (+G), CD27/28 (+C), IVS-I-1 (G-T), IVS-I-5 (G-C), 5'UTR;+40-43 (-AAAC), initiation codon (ATG-AGG), and -30 (T-C)] were genotyped by PCR-based reverse dot-blot (PCR-RDB) (Yaneng Biological Technology Co., Ltd., Shenzhen, China). All procedures were performed in accordance with the manufacturer’s instructions.
Statistical analyses
All statistical analyses were performed using SPSS 22.0 software. Gene mutations, frequency, and spectrum of thalassemia were analyzed by a descriptive method. Given that the obtained data did not conform to normal distribution, hematological parameters data are presented as M (P25, P75) and were compared in different genders and genotypes using the non-parametric Wilcoxons rank sum test. p ≤ 0.05 was considered to be statistically significant.
Results
Prevalence of common thalassemia
Among the 38812 couples, 15231 couples had one or both sides of the couple being positive for thalassemia during screening, and thus, further genetic testing was performed. Among them, 20770 cases were diagnosed with thalassemia, 5862 cases had iron deficiency, while 3830 cases were retained for further analysis. The overall incidence of thalassemia was 26.76% (20770/77624) ((A)), while incidences of α-thalassemia, β-thalassemia, and concurrent α- and β-thalassemia were 17.52% (13596/77624), 6.92% (5375/77624), and 2.32% (1799/77624), respectively ((B)). Moreover, the ratios of α-thalassemia, β-thalassemia, and concurrent α- and β-thalassemia were 65.46%, 25.88%, and 8.66%, respectively ((C)).
Figure 1. (A) The prevalence of common thalassemia; (B) The prevalence of α-thalassemia, β-thalassemia, and concurrent α- and β-thalassemia; (C) The constituent ratio of α-thalassemia, β-thalassemia, and concurrent α- and β-thalassemia.
α-thalassemia genotypes
Twenty-five genotypes of α-thalassemia, which were highly abundant and diverse, were identified and are presented in order of abundance in . Genotype --SEA/αα (46.76%) was the most frequent deletional mutation, followed by −α3.7/αα (20.49%) and −α4.2/αα (7.16%) genotypes. In order of decreasing frequency, the three non-deletional mutations were αCSα/αα (10.54%), αWSα/αα (4.88%), and αQSα/αα (0.94%) genotypes. The top five most frequent double heterozygotes or homozygous genotypes were --SEA/-α3.7 (2.24%), --SEA/αCSα (1.15%), -α3.7/-α3.7 (0.91%), --SEA/-α4.2 (0.88%), and --SEA/αWSα (0.85%).
Table 1. Genotyping of α-thalassemia among people of reproductive age in the border area of Guangxi-Yunnan-Guizhou province, Southwestern China.
β-thalassemia genotypes
Fifteen different genotypes were found in 5375 β-thalassemia subjects. Among them, βCD17/βN and βCD41-42/βN were the dominant genotypes with frequencies of 43.39% and 33.38%, respectively, accounting for the vast majority of β-thalassemia patients (76.77%). In addition, βCD26/βN, βIVS-I-1/βN, βCD71-72/βN, β-28/βN, and βIVS-II-654/βN genotypes accounted for 6.10%, 4.91%, 3.91%, 3.80%, and 2.83% of β-thalassemia patients, respectively. Proportions of the other eight genotypes were all less than 1% ().
Table 2. Genotyping of β-thalassemia among people of reproductive age in the border area of Guangxi-Yunnan-Guizhou province, Southwestern China.
Genotypes of concurrent α- and β-thalassemia
Among the 1799 concurrent mutated α- and β-thalassemia genes, 95 different genotypes were identified. shows the top 35 frequent types, which accounted for 92.50% of all concurrent α- and β-thalassemia genotypes. It was found that --SEA/αα/βCD17/βN was the most prevalent type, accounting for 14.06%. The next five frequent mutations were --SEA/αα/βCD41-42/βN (12.06%), -α3.7/αα/βCD17/βN (10.12%), -α3.7/αα/βCD41-42/βN (9.78%), αCSα/αα/βCD17/βN (5.61%), and αCSα/αα/βCD41-42/βN (4.56%). The remaining 60 genotypes, which all accounted for less than seven cases, are not shown in . Among them, -α3.7/-α3.7/βCD41-42/βN, -α3.7α/αWSα/βCD17/βN, -α4.2/αα/β-28/βN, and αQSα/αα/βCD41-42/βN genotypes were all found in six cases, whereas --SEA/αWSα/βCD17/βN and αCSα/αα/βCD26/βN genotypes were both found in five cases.
Table 3. Genotyping of concurrent α- and β-thalassemia among people of reproductive age in the border area of Guangxi-Yunnan-Guizhou province, Southwestern China.
Forms of thalassemia for which fetuses would be at risk among couples in which both partners were positive for thalassemia isotypes
Among the 38812 couples, couples in which both partners were positive for α-thalassemia isotype were 3414/38812 (8.80%), and those positive for β-thalassemia isotype were 807/38812 (2.08%). The proportion of couples found to be at risk of having a baby with fetal Hb Bart’s disease, HbH disease, HbH or α-thalassemia minor, α-thalassemia minor, and β-thalassemia major or intermedia were 808/38812 (2.08%), 1700/38812 (4.38%), 149/38812 (0.38%), 757/38812 (1.95%), and 807/38812 (2.08%), respectively ().
Table 4. Forms of thalassemia for which fetuses would be at risk among couples in which both partners were positive for thalassemia in isotype among people of reproductive age in the Border Area of Guangxi-Yunnan-Guizhou province, Southwestern China.
Characteristics and hematological parameters of α-thalassemia
Hematological parameters and molecular characteristics of the top 18 frequent α-thalassemia mutations are shown in . Due to the low number of cases, the remaining seven genotypes were not included in the analysis. Among participants of different genders and genotypes, hematological and electrophoretic parameters included MCV (fl), MCH (Pg), Hb (g/dl), HbA2 (%), and HbF (%) are shown in . A significant difference between genders was observed for Hb, with males exhibiting higher Hb levels compared to females (p < 0.001) ((C)). In partial genotypes, MCV and MCH levels were significantly higher in males than in females (p < 0.05) ((A,B)). Interestingly, in a small number of genotypes, females had higher HbA2 and HbF levels than males ((D,E)). shows pairwise comparison results for each parameter between different genotypes. For MCV, MCH, Hb, HbA2, and HbF values, 114, 119, 105, 98, and 7 out of 153 pairwise comparisons were significantly different in males, and 116, 128, 108, 89, and 33 out of 153 pairwise comparisons were significantly different in females, respectively ().
Figure 2. Hematological characterization of α-thalassemia among different genders and genotypes. (A) MCV levels among participants with different genders and genotypes; (B) MCH levels among participants with different genders and genotypes; (C) Hb levels among participants with different genders and genotypes; (D) HbA2 levels among participants with different genders and genotypes; (E) HbF levels among participants with different genders and genotypes. # Males had significantly higher levels (p < 0.05) than females in the same genotypic group, * Males had significantly lower levels (p < 0.05) than females in the same genotypic group. P values for significant differences (p < 0.05) between genotypes of α-thalassaemia are shown. Non-listed comparisons are not significant.
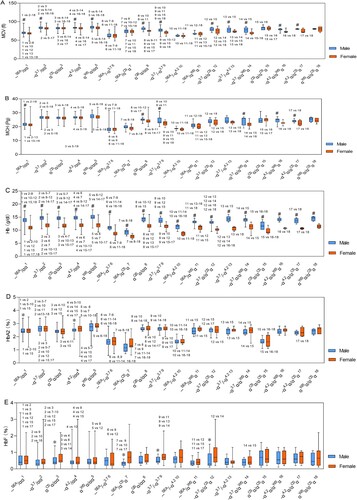
Table 5. Characteristics and hematological parameters of α-thalassemia among people of reproductive age in the border area of Guangxi-Yunnan-Guizhou province, Southwestern China, M (P25, P75).
Characteristics and hematological parameters of β-thalassemia
Characteristics and hematological parameters, including MCV, MCH, Hb, HbA2, and HbF, of the top nine frequent types of β-thalassemia are shown in . Notably, due to the low number of cases, βCD27-28/βN, βCD14-15/βN, βCAP/βN, β-28/βCD17, β-28/βCD41-42, and β-28/βIVS-I-1 were not included in the analysis. The MCV, MCH, Hb, HbA2, and HbF values for participants with different genders and genotypes are shown in . Compared to females, males in each genotypic group had significantly higher Hb levels (p < 0.001) ((C)), significantly higher MCV levels in βCD41-42/βN, βCD26/βN, and β-28/βN genotypic groups (p < 0.05) ((A)), significantly higher MCH levels in β-28/βN and βCD43/βN genotypic groups (p < 0.05) ((B)), and significantly lower HbF levels in βCD17/βN, βCD41-42/βN, βIVS-I-1/βN βCD71-72/βNβ-28/βN, and βIVS-II-654/βN genotypic groups (p < 0.05) ((E)). However, differences in HbA2 levels between genders were not significant. Pairwise comparisons for each parameter between different genotypes are shown in . For MCV, MCH, Hb, HbA2, and HbF values, 24, 27, 19, two, and five out of 36 pairwise comparisons were significantly different in males, and 24, 20, 22, five, and 17 out of 36 pairwise comparisons were significantly different in females, respectively ().
Figure 3. Hematological characterization of β-thalassemia among different genders and genotypes. (A) MCV levels among participants with different genders and genotypes; (B) MCH levels among participants with different genders and genotypes; (C) Hb levels among participants with different genders and genotypes; (D) HbA2 levels among participants with different genders and genotypes; (E) HbF levels among participants with different genders and genotypes. # Males had significantly higher levels (p < 0.05) than females in the same genotypic group, * Males had significantly lower levels (p < 0.05) than females in the same genotypic group. P values for significant differences (p < 0.05) between genotypes of β-thalassaemia are shown. Non-listed comparisons were not significant. The HbA2% level of genotype βCD26/βN was the sum of HbA2% and HbE%, and was not compared with other genotypes.
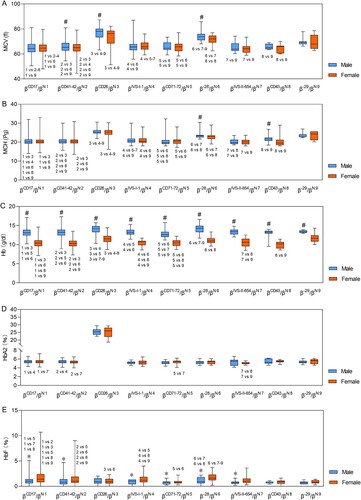
Table 6. Characteristics and hematological parameters of β-thalassemia among people of reproductive age in the border area of Guangxi-Yunnan-Guizhou province, Southwestern China, M (P25, P75).
Characteristics and hematological parameters of concurrent α- and β-thalassemia
Clinical features of the top 10 frequent concurrent α- and β-thalassemia mutations are shown in . The MCV, MCH, Hb, HbA2, and HbF levels for participants from different genders with different genotypes are shown in . Compared to females, males in each genotypic group had significantly higher Hb levels (p < 0.001) ((C)); compared to the α-thalassemia alone group, the concurrent α- and β-thalassemia group had significantly higher HbA2 levels (p < 0.001) ((D)); Compared to the β-thalassemia alone group, the concurrent α- and β-thalassemia group had significantly higher MCV and MCH levels, apart from females with genotype -α3.7/αα/βCD17/βN and both males and females with genotype --SEA/αα/βCD26/βN. Differences in MCV levels for males with genotype -α4.2/αα/βCD17/βN were insignificant. The remaining relationships are illustrated in . Pairwise comparisons for each parameter between different genotypes are shown in . For the MCV, MCH, Hb, HbA2, and HbF values, 16, 15, 14, three, and six out of 45 pairwise comparisons were significantly different in males, and 20, 15, six, 10, and one out of 45 pairwise comparisons were significantly different in females, respectively.
Figure 4. Hematological characterization of concurrent α- and β-thalassemia among different genders and genotypes. (A) MCV levels among participants with different genders and genotypes; (B) MCH levels among participants with different genders and genotypes; (C) Hb levels among participants with different genders and genotypes; (D) HbA2 levels among participants with different genders and genotypes; (E) HbF levels among participants with different genders and genotypes. #Males had significantly higher levels (p < 0.05) than females in the same genotypic group; △Compared to β-thalassemia alone group in the same gender, concurrent α- and β-thalassemia group had significantly higher levels (p < 0.05); ⋆compared to α-thalassemia alone group in the same gender, concurrent α- and β-thalassemia group had significantly higher levels (p < 0.05). ▴Compared to β-thalassemia alone group in the same gender, concurrent α- and β-thalassemia group had significantly lower levels (p < 0.05); ★compared to the α-thalassemia alone group in the same gender, concurrent α- and β-thalassemia group had significantly lower levels (p < 0.05). Non-listed comparisons were not significant. The HbA2% level of genotype --SEA/αα/βCD26/βN was the sum of HbA2% and HbE%, and was not compared with other genotypes except, βCD26/βN.
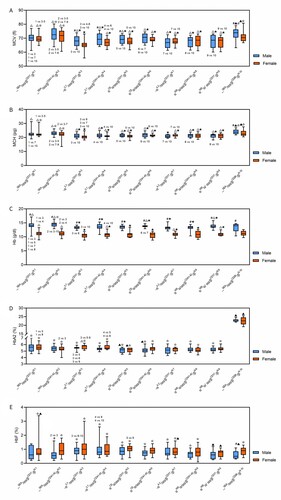
Table 7. Characteristics and hematological parameters of concurrent α- and β-thalassemia among people of reproductive age in the border area of Guangxi-Yunnan-Guizhou province, Southwestern China, M (P25, P75).
Discussion
The high frequency of thalassemia in the border area of Guangxi-Yunnan-Guizhou provinces of southwestern China has become a major public health concern. It is associated with a considerable socio-economic burden. However, currently, data on the prevalence and molecular characterization of thalassemia in this region are not conclusive. The prevalence of thalassemia in this study was higher than that reported in a previous large population-based study conducted in Guangxi, Yunnan, Guizhou, Guangdong, and Hainan provinces [Citation7]. It was also higher than the 24.51% total heterozygote frequency of thalassemia and other haemoglobinopathies reported in the 2010 sampling survey performed in Guangxi province [Citation8]. Moreover, our data showed higher frequencies than those in previous reports [Citation9,Citation10], which may be attributed to the large sample size. Collectively, thalassemia is highly prevalent in the border area of Guangxi-Yunnan-Guizhou provinces, Southwestern China, suggesting that premarital genetic counseling should be performed to prevent the birth of babies with thalassemia major.
We identified 25 α-thalassemia genotypes, including 79.13% deletional mutations, 16.96% non-deletional mutations, and 3.91% concurrent deletional and non-deletional mutations (). The five most frequent α-thalassemia genotypes were --SEA/αα, -α3.7/αα, αCSα/αα, -α4.2/αα, and αWSα/αα genotypes, which accounted for 89.83% of all α-thalassemia genotypes. The spectrum of mutations detected in this study was similar to the findings observed in southern regions of China [Citation9,Citation11,Citation12]. These results form the basis for designing efficient and cost-effective prevention interventions for α-thalassemia major and intermedia.
We also identified 15 different β-thalassemia genotypes, with βCD17/βN, βCD41-42/βN, and βCD26/βN being the three most frequent genotypes, followed by βIVS-I-1/βN, βCD71-72/βN, β-28/βN, and βIVS-II-654/βN. These seven genotypes accounted for 98.32% of all β-thalassemia genotypes. These results indicate a high genetic heterogeneity for β-thalassemia in the border area of Guangxi-Yunnan-Guizhou provinces, Southwestern China. Even though these results differed from the spectrum of β-thalassemia gene mutations in Guangdong, Guangxi, Yunnan, and Guizhou provinces [Citation13–16], the most common genotype (βCD17/βN) was consistent with previous reports [Citation15,Citation16]. Our results indicate a high genetic diversity and significant genetic variations of β-thalassemia in the border area of Guangxi-Yunnan-Guizhou provinces, Southwestern China.
Patients with mild concurrent α- and β-thalassemia mostly exhibited elevated HBA2 and/or HbF levels and had characteristics of β-thalassemia heterozygotes ((D,E)), therefore, the characteristics of α-thalassemia were masked. The high proportions (2.32%) of concurrent α- and β-thalassemia suggests that careful screening of α-thalassemia should be performed in all β-thalassemia patients to obtain accurate genotypes, thereby avoiding misinterpreting the appropriate risk assessment and counseling, especially in prenatal diagnosis [Citation17,Citation18]. In this study, concurrent α- and β-thalassemia that compounded with α+ were associated with significantly low MCV, MCH and Hb levels, compared to α-thalassemia alone group. However, most of the genotypes that combined with α0 had significantly elevated MCV, MCH and Hb levels. Eighteen cases of concurrent α- and β-thalassemia were characterized with intermediate disease phenotypes, including three cases of -α3.7/αα/βCD26/βN, two cases of -α3.7/αα/βIVS-II-654/βN and αCSα/αα/βIVS-I-1/βN respectively, and --SEA/αα/βCD41-42/βN, α3.7/αα/βCD17/βN, -α4.2/αα/βCD17/βN, --SEA/αα/βIVS-I-1/βN, αCSα/αα/βIVS-II-654/βN, etc. other 6 genotypes of one case each. However, the reason is not yet clear. Notably, the β- thalassemia phenotype was not exacerbated in the rest of concurrent α- and β-thalassemia individuals apart from genotype --SEA/αα/βCD26/βN, compared to the β-thalassemia alone group, nearly all genotypes have significant higher value of MCV and MCH, and parts genotypes have significant higher value of Hb. In the same β-thalassemia genotype, individuals with α0 had higher MCV, MCH and Hb levels, relative to those with α+. The MCV, MCH and Hb levels increased with decreasing copy numbers of the functional α-globin gene. This is because, the synthesis of both α and β peptide chains is reduced in concurrent α- and β-thalassemia, and the severity of clinical symptoms in thalassemia patients mainly depend on imbalances of α/β peptide chains [Citation19–21]. Differences in levels of some hematological parameter indicators between concurrent α- and β-thalassemia and α-thalassemia or β-thalassemia alone group were significant. However, it is challenging to strictly distinguish the single types or compound types of thalassemia for the detection of a single sample. Therefore, differentiating between the two cannot be reliably done by using hematological phenotypes alone, it must be based on accurate molecular diagnosis.
A limitation of this study is that we only analyzed common thalassemia. Given the rapid development of molecular diagnostic techniques in recent years, deficiencies and inadequacies of conventional thalassemia gene screening methods have gradually emerged. According to the HbVar Database of Hemoglobin Variants and Thalassemia Mutations (http://globin.bx.psu.edu/hbvar/), as of April 2022, 536 different mutations of thalassemia have been globally characterized. Conventional commercial kits fail to detect rare types of thalassemia, resulting in misdiagnosis. Among the 38812 couples involved in this study, 3830 cases were positive for thalassemia screen but non-iron deficiency, suggesting rare types of thalassemia and/or hemoglobinopathies. From the 3830 cases, we detected 100 samples with rare thalassemia gene mutations using a next-generation sequencing-based (NGS-based) approach ‘Dx0593 thalassemia gene detection (α+β)508plus’ by Shenzhen Huada Gene Co., Ltd. Specifically, 25 cases had rare thalassemia gene mutations, including --THAI/αα, HKαα/-α4.2, -α21.9/αα, HBA2:c.184A > T, SEA-HPFH, β−86/βN, and Chinese Gγ+(Aγδβ)0 among others, while 17 cases had hemoglobinopathies, including HBA2:c.344C > A, HbJ-Wenchang-Wuming, HbG-Coushatta, and Hb G-Honolulu among others. These results suggest that a constant number of rare thalassemias and abnormal hemoglobinopathies exist in the 3830 samples. The NGS-based approach identified both annotated and novel variants, and common and rare mutations in carriers with or without thalassemic trait phenotypes, thereby significantly improving the detection of carrier status, and ultimately decreasing the risk of missed diagnoses and misdiagnoses [Citation7,Citation22–24]. Nevertheless, taking the cost implications and clinical significance into account, screening the priority population for NGS is necessary and there should be economic and policy support. Priority population for NGS testing are: Couples who previously had a child with thalassemia major or intermedia, but either one or both husband and wife were negative for common thalassemia; One of the couples positive for the common thalassemia; Individuals with obvious phenotypes, such as anemia or with abnormal hemoglobin band in hemoglobin analysis but negative for common thalassemia.
Given that there is no effective treatment option for thalassemia, there is an urgent need to prioritize thalassemia prevention and control in mainland China [Citation25,Citation26]. Immediate and concerted actions for thalassemia prevention and control should be mandatory in high prevalence areas [Citation27,Citation28]. Provision of appropriate genetic counselling, prenatal diagnosis and interventions to thalassemia carriers can prevent and control severe thalassemia. In this study, the proportions of couples found to be at risk of having children with thalassemia major and thalassemia intermedia were high (). Therefore, individuals in this high prevalence region should be subjected to premarital thalassemia major and intermedia screening. Since thalassemia intermedia patients have a high genetic heterogeneity and broad phenotypic diversity [Citation29,Citation30], they should be carefully screened for all known disease-causing modifier variants and mutations, with the overarching goal of providing critical clinical information insights into their potential offsprings, and facilitating precise diagnosis and genetic counselling.
Currently, thalassemia diagnosis relies on routine blood examination combined with blood hemoglobin electrophoresis and thalassemia genetic gene detection. Characteristics and hematological parameters have a potential role in thalassemia genotyping [Citation13,Citation31]. The characteristics and hematological parameters in thalassemia patients vary with lifestyle, race, and altitude [Citation32]. Nevertheless, a limited number of studies have described routine blood parameters, electrophoretic detection and quantification for screening thalassemia in the Chinese population. In this study, Hb levels was significantly different between males and females in the same genotype, whether α-thalassemia or β-thalassemia. The Hb reference value for males was higher than that of females. Pregnant women, especially those with thalassemia, were more likely to be anaemic, the reasons and mechanisms, however, should be further investigated. In our study, pregnant women accounted for the majority of females. These may explain why males had significantly higher Hb levels, relative to females with the same genotype. Furthermore, males and females with partial genotypes had different MCV, MCH, HbA2, and HbF levels. The higher Hb levels in males were comparable to those found by Zhang et al. [Citation32]. But, differences in MCV, MCH, HbA2, and HbF levels between males and females were inconsistent with results reported by Zhang et al. [Citation32]. In this study, the prevalence of βCD26/βN was 0.42% (328/77624). However, HPLC could not differentiate between HbA2 and HbE because they had the same elution time, hence, capillary electrophoresis techniques are recommended in future studies. Our findings form the basis for future studies on thalassemia.
In summary, this large-scale analysis of the prevalence and molecular characterization of common thalassemia provides valuable knowledge on thalassemia in China. Thalassemia is highly prevalent in the border area of Guangxi-Yunnan-Guizhou provinces, Southwestern China, and the thalassemia genotypes are characterized by a huge genetic diversity. Collectively, these findings provide valuable baseline data for prenatal diagnosis and genetic counseling in the region.
Institutional review board statement
This study was approved by the Affiliated Hospital of Youjiang Medical University for Nationalities, Baise, China.
Data sharing statement
Readers can get the data of this paper by contacting us via E-mail: 1123049646 @qq.com.
ARRIVE guidelines statement
The authors have read the ARRIVE guidelines, and the manuscript was prepared and revised according to the ARRIVE guidelines.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Birgens H, Ljung R. The thalassaemia syndromes. Scand J Clin Lab Invest. 2007;67(1):11–25.
- Shafique F, Ali S, Almansouri T, et al. Thalassemia, a human blood disorder. Braz J Biol. 2021;83:e246062.
- Martin M, Haines D. Clinical management of patients with thalassemia syndromes. Clin J Oncol Nurs. 2016;20(3):310–317.
- Jiang F, Zuo L, Li J, et al. Evaluation of intervention strategy of thalassemia for couples of childbearing ages in centre of southern China. J Clin Lab Anal. 2021;35(10):e23990.
- Ip HW, So CC. Diagnosis and prevention of thalassemia. Crit Rev Clin Lab Sci. 2013;50(6):125–141.
- Li CK. New trend in the epidemiology of thalassaemia. Best Pract Res Clin Obstet Gynaecol. 2017;39:16–26.
- Shang X, Peng Z, Ye Y, et al. Rapid targeted next-generation sequencing platform for molecular screening and clinical genotyping in subjects with hemoglobinopathies. EBioMedicine. 2017;23:150–159.
- Xiong F, Sun M, Zhang X, et al. Molecular epidemiological survey of haemoglobinopathies in the Guangxi Zhuang Autonomous Region of southern China. Clin Genet. 2010;78(2):139–148.
- He S, Qin Q, Yi S, et al. Prevalence and genetic analysis of alpha- and beta-thalassemia in Baise region, a multi-ethnic region in southern China. Gene. 2017;619:71–75.
- Pan HF, Long GF, Li Q, et al. Current status of thalassemia in minority populations in Guangxi, China. Clin Genet. 2007;71(5):419–426.
- Long J, Pang W, Sun L, et al. Diagnosis of a family with the novel -alpha(21.9) thalassemia deletion. Hemoglobin. 2015;39(6):419–422.
- Tang W, Zhang C, Lu F, et al. Spectrum of alpha-thalassemia and beta-thalassemia mutations in the Guilin region of southern China. Clin Biochem. 2015;48(16-17):1068–1072.
- Gu H, Wang YX, Du MX, et al. Effectiveness of using mean corpuscular volume and mean corpuscular hemoglobin for beta-thalassemia carrier screening in the Guangdong population of China. Biomed Environ Sci. 2021;34(8):667–671.
- He S, Li D, Lai Y, et al. Prenatal diagnosis of beta-thalassemia in Guangxi Zhuang autonomous region, China. Arch Gynecol Obstet. 2014;289(1):61–65.
- Huang TL, Zhang TY, Song CY, et al. Gene mutation spectrum of thalassemia among children in Yunnan province. Front Pediatr. 2020;8:159.
- Huang SW, Liu XM, Li GF, et al. Spectrum of beta-thalassemia mutations in Guizhou province, PR China, including first observation of codon 121 (GAA > TAA) in Chinese population. Clin Biochem. 2013;46(18):1865–1868.
- Yamsri S, Sanchaisuriya K, Fucharoen G, et al. Prevention of severe thalassemia in northeast Thailand: 16 years of experience at a single university center. Prenat Diagn. 2010;30(6):540–546.
- Rahim F, Kaikhaei B, Zandian K, et al. Co-inheritance of alpha-and beta-thalassemia in Khuzestan province, Iran. Hematology. 2008;13(1):59–64.
- Wee YC, Tan KL, Kuldip K, et al. Alpha-thalassaemia in association with beta-thalassaemia patients in Malaysia: a study on the co-inheritance of both disorders. Community Genet. 2008;11(3):129–134.
- Shang X, Xu X. Update in the genetics of thalassemia: what clinicians need to know. Best Pract Res Clin Obstet Gynaecol. 2017;39:3–15.
- Mettananda S, Higgs DR. Molecular basis and genetic modifiers of thalassemia. Hematol Oncol Clin North Am. 2018;32(2):177–191.
- Zhang H, Li C, Li J, et al. Next-generation sequencing improves molecular epidemiological characterization of thalassemia in Chenzhou region. P.R. China. J Clin Lab Anal. 2019;33(4):e22845.
- He J, Song W, Yang J, et al. Next-generation sequencing improves thalassemia carrier screening among premarital adults in a high prevalence population: the Dai nationality, China. Genet Med. 2017;19(9):1022–1031.
- Chen P, Yu X, Huang H, et al. Evaluation of Ion Torrent next-generation sequencing for thalassemia diagnosis. J Int Med Res. 2020;48(12):1–9.
- Jiang F, Chen GL, Li J, et al. Pre gestational thalassemia screening in Mainland China: the first two years of a preventive program. Hemoglobin. 2017;41(4-6):248–253.
- He P, Yang Y, Li R, et al. Prenatal control of Hb Bart's disease in mainland China: can we do better? Hemoglobin. 2014;38(6):435–439.
- Weatherall DJ. The inherited diseases of hemoglobin are an emerging global health burden. Blood. 2010;115(22):4331–4336.
- Weatherall D. The inherited disorders of haemoglobin: an increasingly neglected global health burden. Indian J Med Res. 2011;134(4):493–497.
- Lal A, Goldrich ML, Haines DA, et al. Heterogeneity of hemoglobin H disease in childhood. N Engl J Med. 2011;364(8):710–718.
- Piga A, Perrotta S, Gamberini MR, et al. Luspatercept improves hemoglobin levels and blood transfusion requirements in a study of patients with beta-thalassemia. Blood. 2019;133(12):1279–1289.
- Zhan W, Guo H, Hu S, et al. Comparison of cord blood hematological parameters among normal, alpha-thalassemia, and beta-thalassemia fetuses between 17 and 38 weeks of gestation. Sci Rep. 2021;11(1):3844.
- Zhang J, He J, Mao X, et al. Haematological and electrophoretic characterisation of beta-thalassaemia in Yunnan province of Southwestern China. BMJ Open. 2017;7(1):e013367.