
ABSTRACT
Objectives
To identify the clinical phenotypic and molecular pathogeneses of four cases of coagulation factor XII deficiency and to deepen the cognition of this disease.
Methods
Coagulation tests were performed through one stage of coagulation on a STAGO coagulation analyser. Coagulation factor XII antigen was detected using enzyme-linked immunosorbent assay. The species conservatism and structural change of mutant proteins were analysed using MegAlign and PYMOL. Meanwhile, missense variants and a novel splice site variant were identified using PolyPhen2 and NetGene2.
Results
The four cases had an observably prolonged activated partial thromboplastin time but without obvious bleeding tendency. Their coagulation factor XII activity (FⅫ:C) and antigen (FXII:Ag) were greatly reduced. Six mutations were detected: NM_000505.4:c.398-1G>A, NP_000496.2:p.(Pro182Leu), NP_000496.2:p.(Ser479Ter), NP_000496.2:p.(Cys559Arg), NC_000005.10:g.7217_7221delinsGTCTA and NM_000505.4:c.1681-1G>A. The first five are newly discovered mutations. The two missense mutation sites were highly conservative, and their protein secondary structure changes may occur not only on the mutation sites but also on other domains. In silico analysis revealed that NP_000496.2:p.(Pro182Leu) may be BENIGN, NP_000496.2:p.(Cys559Arg) may be damaging, and that NM_000505.4:c.398-1G>A and NM_000505.4:c.1681-1G>A are crucial for splicing.
Conclusion
We found six types of mutations, of which five were novel. The two missense mutation sites might be closely related to the function of coagulation factor XII. The mutations were the primary culprits of factor XII deficiency.
Introduction
Human coagulation factor XII (FXII; F12) is a single-chain glycoprotein synthesized by hepatocytes and secreted to plasma as a serine protease zymogen [Citation1]. F12 is in 5q33-qter of chromosomal band, covering 14 exons and 13 introns [Citation2]. Mature FXII is composed of 596 amino acids, containing an NH2-terminal heavy chain and a COOH-terminal light chain [Citation3]. These two chains are cleaved at Arg353-Val354 when activated. The heavy chain incorporates six domains, including fibronectin type II domain, two epidermal growth factor-like domains, fibronectin type I domain, the Kringle domain and pro-rich region, each of which performs a different function. In addition, the light chain is a catalytic domain whose active site is composed of His394, Asp442 and Ser544 [Citation2].
FXII deficiency is a rare and autosomal recessive genetic disease whose incidence is close to 1/1,000,000. FXII is the initiator of the intrinsic coagulation pathway [Citation4], but most individuals with FXII deficiency have no bleeding tendency [Citation5,Citation6], which may lead to the low detection ratio of FXII deficiency. Therefore, the molecular mechanism of FXII deficiency needs further understanding. Only approximately 53 types of gene mutation are included in the Human Gene Mutation Database for FXII deficiency, possibly because of the low incidence of the disease or its lack of clinical manifestations. In this study, we analysed four cases of FXII deficiency and found six types of gene mutation, of which five were novel.
Materials and methods
Identification of study subjects
Routine coagulation tests in the clinical laboratory of China include plasma prothrombin time, activated partial thromboplastin time (APTT), thrombin time and fibrinogen. For test samples with prolonged APTT, blood coagulation factor Ⅷ, Ⅸ, Ⅺ and Ⅻ activities (FⅧ:C, FⅨ:C, FⅪ:C, FⅫ:C) were determined and APTT plasma mixing study was performed, followed by factor Ⅻ antigen (FXII:Ag) if FⅫ:C was abnormal and APTT plasma mixing study could be rectified. In samples with prolonged APTT, rectified APTT plasma mixing study and reduced FⅫ:C and FXII:Ag, we identified potential subjects from 398,320 patients in our hospital from 2009 to 2017.
Subject
Case 1 was a 2-year-old Chinese boy who presented with scrotal emptiness but without a history of abnormal bleeding. Case 2 was a 33-year-old Chinese pregnant woman who was hospitalized for a small amount of colporrhagia without abdominal pain. Case 3 was a 29-year-old man whose check-up showed that he was in good health. Case 4, whose parents were inbred, was a 23-year-old woman diagnosed with angioimmunoblastic T-cell lymphoma. They had no history of abnormal bleeding and no abnormal liver and renal functions, but coagulation teats showed that APTT was obviously prolonged, APTT plasma mixing study could be rectified and FⅫ: C and FXII:Ag reduced clearly. These patients were diagnosed with CRM-negative heterozygous or homozygous FXII deficiency.
Normal control
One hundred healthy individuals (50 men and 50 women) aged 18–50 without abnormal liver and renal functions and without history of abnormal bleeding, thrombus and taking antithrombotic drugs served as normal controls.
Reagents and instruments
PT, APTT, FIB, TT, FⅧ: C, FⅨ: C, FⅪ: C and FⅫ: C were detected through one stage of coagulation on a STAGO coagulation analyser. Enzyme-linked immunosorbent assay was used to detect FXII:Ag. Mammalian genomic DNA miniprep kit and primers were purchased from ASSAYPRO (American), Axygen (Hangzhou, China) and Sangon Biotech (Shanghai, China).
Primer design
On the basis of F12, we designed seven pairs of primers to cover 14 exons, partial introns and Factor Ⅻ flanking sequence by primer 5.0TM. provides information on the primer sequences, the length of amplification products and annealing temperature.
Table 1. Primer sequences, the length of amplification products and annealing temperature.
Polymerase chain reaction (PCR)
The F12 fragments were amplified using PCR. Peripheral blood samples of 4 cases and 100 normal controls were prepared to obtain genomic DNA as a template. Then, 1 μL of forward primer, 1 μL of reverse primer, 3 μL of template, 25 μL of Taq PCR MasterMix and 20 μL of double distilled water were mixed into the reaction system. Hot-start PCR reactions were performed as follows: 30 cycles of denaturation at 94°C for 1 min, annealing at annealing temperature () for 40 s and polymerization at 72°C for 5 min. The concentration of the primers was 10 μM and prepared with a final concentration of 0.2 μM.
Gene analysis
F12 of four cases and Case 4’s parents was amplified. The PCR products were purified and analysed by Shanghai Sunny Biotechnology Co., Ltd., and potential mutation sites were identified by comparison with the NCBI F12 reference sequence KP123628.1. If we did not find the potential mutation site in 100 normal controls and in the SNP database (SNP linked to Gene (geneID:2161) Via Contig Annotation (nih.gov); last visit December 2021), we identified it as the mutation site.
Sequence conservatism and protein structure
Sequence conservatism and protein structure were analysed using MegAlign and PYMOL.
Results
Coagulation test results
The results of coagulation test showed that all four cases had an observably prolonged APTT and that their FⅫ activity and antigen were greatly reduced ().
Table 2. Hemostatic laboratory results.
Screening of mutation site
and show the mutations in F12 of the 4 cases. Cases 1, 2 and 3 were heterozygous mutations, whereas case 4 was a homozygous mutation. Moreover, both parents of Case 4 were heterozygous with NP_000496.2:p.(Cys559Arg).
Figure 1. Information of mutant FXII DNA. (a1) NM_000505.4:c.398-1G > A in case 1. (a2) NC_000005.10:g.7217_7221delinsGTCTA in case 1. (b1) NM_000505.4:c.398-1G > A in case 2. (b2) NP_000496.2:p.(Ser479Ter) in case 2. (c1) NP_000496.2:p.(Pro182Leu) in case 3. (c2) NM_000505.4:c.1681-1G > A in case 3. (d1) NP_000496.2:p.(Cys559Arg) in case 4. (n) showed normal.
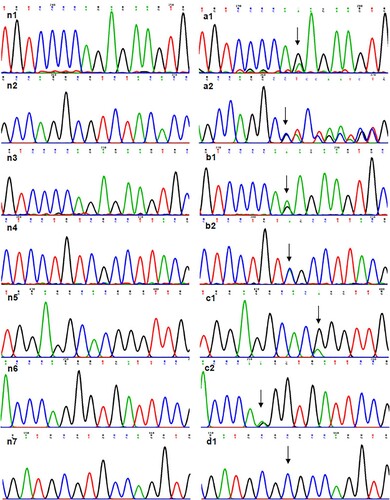
Table 3. Screening of the mutation site.
Conserved analysis of missense mutation site
Sequence analysis of F12 revealed two missense mutations, NP_000496.2:p.(Pro182Leu) (case3) and NP_000496.2:p.(Cys559Arg) (case4). These two points were analysed for amino acid conservatism of FXII protein. In , red, orange, green, baby blue and mazarine meant high to low conservatism. Both 182 P-FXII and 559 C-FXII were red, indicating that they were highly conservative.
Figure 2. Conserved analysis of missense mutation site. (a) 182 P- FXII’s conservatism as the black arrow showed. (b) 559 C- FXII’s conservatism as the black arrow showed.
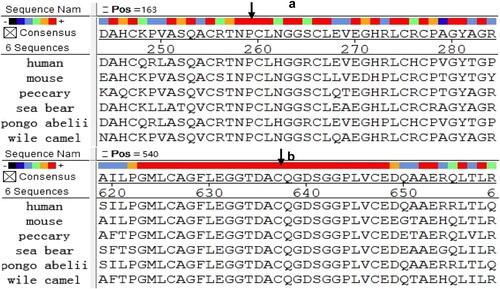
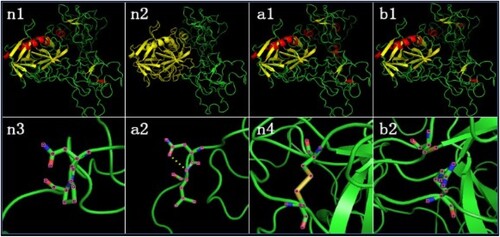
Figure 3. Predictive analytics of FXII protein structure. (n1) Wild-type FXII protein’s cartoon model n2 In wild-type FXII protein’s cartoon model, yellow represents light chain and green represents heavy chain. (a1) Mutant FXII (NP_000496.2:p.(Pro182Leu)) protein’s cartoon model. (b1) Mutant FXII (NP_000496.2:p.(Cys559Arg)) protein’s cartoon model. (n3) Local structure model of 181 N- FXII and 182 P- FXII in wild-type FXII protein. (a2) 162 N- FXII and 182 L- FXII built hydrogen bond in mutant FXII (NP_000496.2:p.(Pro182Leu)) protein. (n4) Disulfide bond between 559-C FXII and 590-C FXII in wild-type FXII protein. (b2) Disulfide bond between 559 and 590 disappeared in mutant FXII (NP_000496.2:p.(Cys559Arg)) protein.
Predictive analytics of FXII protein structure
Results of PYMOL showed that two missense mutations caused secondary protein structure change. As a result of NP_000496.2:p.(Pro182Leu), 182-L-FXII and 181-N-FXII formed hydrogen bonds absent in wild-type FXII protein, and 152→156 (RTEQA) and 193→198(EGHRLC) formed two new α-helixes. As a result of NP_000496.2:p.(Cys559Arg), the disulphide bond between 559 and 590 that existed in wild-type FXII protein disappeared; 77→80 (PNFD), 296→299 (QTPT), 343→346 (AKRE) and 444→456 (SPV) formed four new α-helixes; and 564→571 (GGPLVCED) replaced 564→570(GGPLVCE) to form a new β-sheet ().
In silico analysis of missense variants and splice site variant
PolyPhen2 predicted NP_000496.2:p.(Pro182Leu) to be benign with a score of 0.132 (sensitivity 0.93; specificity 0.86) and NP_000496.2:p.(Cys559Arg) to be probably damaging with a score of 1.000 (sensitivity: 0.00; specificity: 1.00). NetGene2-2.42 predicted NM_000505.4:c.398-1G > A and NM_000505.4:c.1681-1G > A to have confidence scores of 0.8 and 0.96 by NetGene2-2.42, which strongly suggest they are intron splice sites. The two intron splice sites disappeared when the mutated sequence was used for prediction.
Discussion
In the study, four FXII deficiency cases whose APTT was obviously prolonged had no obvious bleeding tendency. This result supports that FXII deficiency is not associated with excessive bleeding [Citation7] and that FXII is more like a bystander in haemostasis [Citation5,Citation8,Citation9]. Although FXII serves as an activator in the intrinsic pathway, FXI can initiate coagulation via the TF-FVIIα complex, which is produced by the extrinsic pathway [Citation10–12]. Under physiological conditions, the blood vessel would release tissue factors when it was damaged. The latter participated in the formation of the TF-FVIIα complex, which could start the extrinsic pathway. In addition, a small amount of thrombin as a product of the extrinsic pathway could activate factor XI to start the intrinsic pathway [Citation12,Citation13]. In the APTT test, the intrinsic pathway was started by FXIIα, which was activated by kaolin and diatomite. The above factors might explain why the person had no obvious bleeding tendency when his APTT prolonged greatly. Moreover, APTT plasma mixing study of the four cases could be rectified, which could eliminate the possibility of the appearance of FXII inhibitor. This finding supports that the lack of FXII:C and FXII:Ag is the main reason for prolonged APTT.
Gene analysis revealed six mutations, of which five were novel, including two splicing mutations, one nonsense mutation, one short fragment deletion and two missense mutations. One of the splicing mutations was NM_000505.4:c.398-1G > A located in the splicing site, which led to breaking the GT-AG rule. Furthermore, it is located in the middle of the heavy chain, which may cause a serious influence. Splicing mutations were reported rarely, as shown in the Human Gene Mutation Database. Another one was NM_000505.4:c.1681-1G > A, which was reported by Schloesser [Citation14]. Analysis of the potential splice variant indicated that these two sites play a decisive role in the splicing of corresponding sites. If the prediction is correct, they will form completely different proteins or cause them to degrade.
The nonsense mutation NP_000496.2:p.(Ser479Ter) caused the early termination of translation. The truncated segment lost the substrate-binding zone and activation zone. Three nonsense mutations whose position was far away from NP_000496.2:p.(Ser479Ter) were reported in the HGMD database, but their NMD was not studied. Suzuki K[Citation15] showed that most 346 N-FXII is degraded intracellularly through endoplasmic reticulum-associated degradation as the protein quality control system, resulting in an insufficient secretion phenotype. However, the changes in the metabolism of the mutant protein cannot be determined from the current experimental data.
The two missense mutations were NP_000496.2:p.(Pro182Leu) in the heavy chain and NP_000496.2:p.(Cys559Arg) in the light chain. These two missense mutations played an essential role in the great reduction of FXII activity and antigen in two cases. This result coincided with other reported missense mutations [Citation16–18]. Some researchers constructed site-directed mutagenesis and expression vectors to study the FXII expression and secretion in wild and mutant types. Even point mutation might impact its expression and secretion [Citation19–21]. Then, we focused on the conserved sequence and protein structure in the mutation point. Results showed that 182-P FXII and 559-C FXII were highly conservative. This result suggests that they have an intimate connection with the metabolism and function of FXII. Meanwhile, these two missense mutations caused the change in FXII secondary structure, not just around the mutation site. Thus, it might become the chief culprit of FXII deficiency. Cys559 to Arg led to disulphide bond disappearing between 559-C FXII and 590-C FXII. Miyata T [Citation22] reported that Cys590 to Ser results in a conformational change, interfering with substrate binding, the same deletion with FXII activity and antigen greatly reducing. It is powerful evidence to embody the importance of point mutation. PYMOL showed that Cys559 to Arg caused structural changes of 96→99 (PNFD), 315→318 (QTPT) and 583→590 (GGPLVCED). They are located in the fibronectin type II, Kringle domain and light chain, which might cause functional abnormalities in the corresponding area. The HGMD database (http://www.hgmd.cf.ac.uk/ac/index.php; last visit December 2021) showed that the proportion of light chain mutations was more than half. In addition, the mutations were associated with FXII deficiency [Citation23–25]. This finding is consistent with the result of the conserved analysis that the region was mostly a highly conservative sequence. Meanwhile, the software showed that NP_000496.2:p.(Pro182Leu) led 182-L-FXII and 181-N-FXII to form hydrogen bonds, which may damage the natural space conformation of the protein, whereas 171→175 (RTEQA) and 212→217(EGHRLC) formed new α-helixes. They are located in fibronectin type I and epidermal growth factor-like domains, which might impact the function of these domains. This finding is consistent with the prediction of PolyPhen2.
In the study, we found six types of mutation, of which five were novel, which contributed to the pathogenesis of factor XII deficiency. However, the specific changes about the structure, function and metabolic processes of mutant proteins and their clinical significance warrant further study.
Acknowledgments
Given the limited number of references allowed in this perspective, we wish to thank the colleagues who are not specifically cited for their contribution and their understanding.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
Notes on contributors
Shanshan Li
Shanshan Li works at the Medical Laboratory Center of The Second Affiliated Hospital of Wenzhou Medical University. Their research interests include thrombosis and hemostasis. Shanshan Li’s study mainly involves FXII.
Kuangyi Shu
Kuangyi Shu works at the Medical Laboratory Center of The Second Affiliated Hospital of Wenzhou Medical University. Their research interests include thrombosis and hemostasis.
Fanfan Li
Fanfan Li is a Ph.D. candidate, whose research focuses on thrombosis.
Xiao Yang
Xiao Yang has a master’s degree. His research interests include thrombosis, hemostasis and schizophrenia; and works in Wenzhou Seventh People’s Hospital.
Wei Yang
Wei Yang is a Ph.D. candidate at the School of Laboratory Medicine and Life Science, Wenzhou Medical University. His research interests include thrombosis and hemostasis, and tumor-educated platelets.
Manli Ye
Manli Ye works at The Medical Laboratory Center of the Second Affiliated Hospital of Wenzhou Medical University. Their research interests include thrombosis and hemostasis.
Xiaoou Wang
Xiaoou Wang works at the Medical Laboratory Center of The Second Affiliated Hospital of Wenzhou Medical University. Their research interests include thrombosis and hemostasis.
Minghua Jiang
Minghua Jiang is a Director of Laboratory Medicine and Medical Laboratory Center of The Second Affiliated Hospital of Wenzhou Medical University. His research interests include thrombosis and hemostasis, abnormal platelet function, and tumor-educated platelets.
References
- Tans G, Rosing J. Structural and functional characterization of factor XII. Semin Thromb Hemost. 1987;13:1–14. doi:10.1055/s-2007-1003471.
- Cool DE, MacGillivray RT. Characterization of the human blood coagulation factor XII gene. intron/exon gene organization and analysis of the 5'-flanking region. J Biol Chem. 1987;262:13662–13673. doi:10.1055/s-0038-1642800.
- McMullen BA, Fujikawa K. Amino acid sequence of the heavy chain of human alpha-factor XIIa (activated Hageman factor). J Biol Chem. 1985;260:5328–5341. doi:10.1016/0165-022X(85)90070-3.
- Renne T, Schmaier AH, Nickel KF, et al. In vivo roles of factor XII. Blood. 2012;120:4296–4303. doi:10.1182/blood-2012-07-292094.
- Jingxuan L CCB, Akin A, James B, et al. Knockdown of liver-derived factor XII by GalNAc-siRNA ALN-F12 prevents thrombosis in mice without impacting hemostatic function. Thromb Res. 2020;196:200–205. doi:10.1016/j.thromres.2020.08.040.
- Fernandes HD, Newton S, Rodrigues JM. Factor XII deficiency mimicking bleeding diathesis: A unique presentation and diagnostic pitfall. Cureus. 2018;10:2817, doi:10.7759/cureus.2817.
- Mirjam B, Christian N, Tobias H, et al. Influence of factor XII deficiency on activated partial thromboplastin time (APTT) in critically ill patients. J Thromb Thrombolysis. 2019;48:466–474. doi:10.1007/s11239-019-01879-w.
- Juang LJ, Mazinani N, Novakowski SK, et al. Coagulation factor XII contributes to hemostasis when activated by soil in wounds. Blood Adv. 2020;4:1737–1745. doi:10.1182/bloodadvances.2019000425.
- Johansson K, Johansson L, Nilsson TK, et al. Factor XII concentrations and risk of intracerebral haemorrhage. A prospective case-referent study. J Stroke Cerebrovasc Dis. 2021;30:105565, doi:10.1016/j.jstrokecerebrovasdis.2020.105565.
- Puy C, Rigg RA, McCarty OJ. The hemostatic role of factor XI. Thromb Res. 2016;141(Suppl 2):S8–S11. doi:10.1016/S0049-3848(16)30354-1.
- Maroney SA, Cooley BC, Ferrel JP, et al. Absence of hematopoietic tissue factor pathway inhibitor mitigates bleeding in mice with hemophilia. Proc Natl Acad Sci U S A. 2012;109:3927–3931. doi:10.1073/pnas.1119858109.
- Zucker M, Seligsohn U, Salomon O, et al. Abnormal plasma clot structure and stability distinguish bleeding risk in patients with severe factor XI deficiency. J Thromb Haemost. 2014;12:1121–1130. doi:10.1111/jth.12600.
- He R, Chen D, He S. Factor XI: hemostasis, thrombosis, and antithrombosis. Thromb Res. 2012;129:541–550. doi:10.1016/j.thromres.2011.11.051.
- Schloesser M, Hofferbert S, Bartz U, et al. The novel acceptor splice site mutation 11396(G–>A) in the factor XII gene causes a truncated transcript in cross-reacting material negative patients. Hum Mol Genet. 1995;4:1235–1237. doi:10.1093/hmg/4.7.1235.
- Suzuki K, Murai K, Suwabe A, et al. Factor XII Ofunato: Lys346Asn mutation associated with blood coagulation factor XII deficiency causes impaired secretion through a proteasome-mediated degradation. Thromb Res. 2010;125(5):438–443. doi:10.1016/j.thromres.2009.12.004.
- Liu M, Wang H, Lin M, et al. A novel homozygous missense mutation (Met527Ile) in a consanguineous marriage family with inherited factor XII deficiency. Hematology. 2020;25(1):502–506. doi:10.1080/16078454.2020.1859249.
- Wang Y, Zhang H, Liu S, et al. Double heterozygous mutations (Cys247Tyr and 252delAsn) cause factor XII deficiency in a Chinese family. Hamostaseologie. 2020;40:650–654. doi:10.1055/a-1181-0390.
- Zhang H, Liu S, Lin C, et al. Compound heterozygous mutations Glu502Lys and Met527Thr of the FXII gene in a patient with factor XII deficiency. Hematology. 2019;24:420–425. doi:10.1080/16078454.2019.1598679.
- Kondo S, Tokunaga F, Kawano S, et al. Factor XII tenri, a novel cross-reacting material negative factor XII deficiency, occurs through a proteasome-mediated degradation. Blood. 1999;93:4300–4308. doi:10.1016/S0887-7963(99)80035-2.
- Oguchi S, Ishii K, Moriki T, et al. Factor XII shizuoka, a novel mutation (Ala392Thr) identified and characterized in a patient with congenital coagulation factor XII deficiency. Thromb Res. 2005;115:191–197. doi:10.1016/j.thromres.2004.08.027.
- Iijima K, Arakawa Y, Sugahara Y, et al. Factor XII Osaka: abnormal factor XII with partially defective prekallikrein cleavage activity. Thromb Haemostasis. 2011;105:473–478. doi:10.1160/TH10-02-0123.
- Miyata T, Kawabata S, Iwanaga S, et al. Coagulation factor XII (Hageman factor) Washington D.C.: inactive factor XIIa results from Cys-571-Ser substitution. Proc Natl Acad Sci U S A. 1989;86:8319–8322. doi:10.1073/pnas.86.21.8319.
- Subramaniam S, Thielmann I, Morowski M, et al. Defective thrombus formation in mice lacking endogenous factor VII activating protease (FSAP). Thromb Haemostasis. 2015;113:870–880. doi:10.1160/TH14-06-0519.
- Cheng Q, Tucker EI. Pine MS, et al. A role for factor XIIa-mediated factor XI activation in thrombus formation in vivo. Blood. 2010;116:3981–3989. doi:10.1182/blood-2010-02-270918.
- Hopp S, Albert-Weissenberger C, Mencl S, et al. Targeting coagulation factor XII as a novel therapeutic option in brain trauma. Ann Neurol. 2016;79:970–982. doi:10.1002/ana.24655.