
ABSTRACT
Objectives
To report the hematolgocial and molecular features of a nove β-globin variant in a Chinese fimaly.
Methods
The proband was a 19-year-old Chinese man whose Hb analysis by HPLC for thalassemia revealed an abnormal peak. Hb analysis was performed by HPLC and CE. Gap-PCR and PCR-reverse dot blot hybridization (PCR-RDB) were used to detect the common mutations in Chinese population. DNA sequencing was used to determine the Hb variant.
Results
The Hb variant and Hb A can be separated but co-elutes with Hb F by the CE method. However, the variant can be separated from Hb A0, Hb F, and Hb A2 using HPLC. DNA sequencing showed a mutation of codon 3 in the β-globin gene. His wife’s HPLC revealed a high value of Hb A2, which proved to be the Hb E using PCR-RDB.
Conclusion
It was the first report of the mutation, so we named it Hb Jiangnan according to the place of residence of the proband. It can be separated by HPLC but not CE. Hb Jiangnan can cause an increased level of Hb A2.
Introduction
Hemoglobinopathies (including thalassemias and hemoglobin variants) are the most common inherited monogenic disorders, characterized by the abnormal structure of one or more globin chains [1, 2]. Up to date, more than 1800 kinds of hemoglobinopathies have been registered in two databases (https://globin.bx.psu.edu/cgi-bin/hbvar/counter and https://www.ithanet.eu/), of Which more than 1300 hemoglobin (Hb) variants. Most of these do not have the hematological or clinical phenotype and are often detected incidentally during thalassemia screening, Hb A1c analysis, or newborn screening [3–5]. Although most Hb variants heterozygotes are asymptomatic, compound heterozygosity of thalassemia and Hb variant produce significant clinical symptoms. The common example is that the combination of Hb E and β-thalassemia can cause thalassemia intermedia, even thalassemia major [6,7]. Therefore, detection of the Hb variant may have important significance for genetic counseling and prenatal diagnosis, especially novel Hb variants. In this study, we report a novel β-globin variant patient with an increasing level of Hb A2 but normal hematological parameters.
Materials and methods
Subjects and samples
The proband, a 19-year-old man, was from Nanning city of Guangxi Zhuang Autonomous Region. He was recommended to visit Jiangbin Hospital of Guangxi Zhuang Autonomous Region for a routine pregnancy examination with his wife. Thalassemia is currently the most common autosomal recessive blood disorder in Guangxi, so this couple was screened on the doctor's advice for thalassemia. With the couple's consent, we obtained samples of their venous blood and transported them to the People’s Hospital of Guangxi Zhuang Autonomous Region for testing.
Hematological analysis
Blood samples were collected in EDTA anticoagulant from the couple, and hematological indexes were determined using an automated cell counter (DXH800; Beckman Coulter, Kraemer Boulevard Brea, CA, USA). Hemoglobin fractions were separated using high-performance liquid chromatography (HPLC) with the β-thalassemia Short Program (VARIANT ⅡTM; Bio-Rad, Hercules, CA, USA). Compared with HPLC, capillary electrophoresis was analyzed following the manufacturer’s guidelines (Capillarys 2 Flex Piercing; Sebia, Lisses, Paris, France).
Routine genetic analysis
According to our region's prevention policy, routine thalassemia genetic analysis is required if the screening results are positive. Gap-polymerase chain reaction (Gap-PCR) was used to detect the deletions of α-thalassemia (including –SEA, –THAI, -α3.7, -α4.2) with thalassemia analysis kits (Yishengtang, Shenzhen, Guangdong, China). The PCR and reverse dot blot hybridization was utilized to detect the common non-deletions of α-thalassemia [including Hb Constant Spring (Hb CS, HBA2:c.427T > C), Hb Quong Sze (Hb QS, HBA2: c.377T > C), Hb Westmead (Hb WS, HBA2: c.369C > G)]. PCR and reverse dot blot hybridization was also used to detect for 17 common β-thalassemia mutations: −32 (C>A), −30 (T>C), −29 (A>G), −28 (A>G), codons 14/15 (+G), codon 17 (A>T), codon 26 (G>A) (Hb E), codons 27/28 (+C), codon 31 (-C), codons 41/42 (-TCTT), codon 43 (G>T), codons 71/72 (+A), IVS-Ⅰ−1 (G>T), IVS-Ⅰ−5 (G>C), IVS-Ⅱ−654 (C>T), Cap+1 (A>C) and initiation codon (ATG>AGG).
DNA sequencing
DNA sequencing was performed using a 3500 XL genetic analyzer sequencer (Applied Biosystems, Foster City, CA, USA). As we described in the previous article [8], the amplification conditions and primers of the HBB gene were used in this study.
Results
His hematological indexes were as follows: hemoglobin (Hb) 139 g/L (reference: 120-160 g/L), mean corpuscular volume (MCV) 87.4 fL (reference: 80.0-99.0 fL), mean corpuscular hemoglobin (MCH) 28.7 pg (reference: 27.0-35.0 pg). His wife complete blood count (CBC) was Hb 97 g/L, MCV 76.8 fL, MCH 25.3 pg ().
Table 1. The hematological and molecular data of the family.
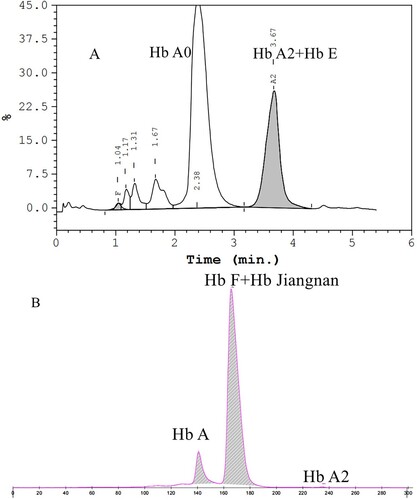
Hemoglobin separation was first performed by HPLC. It showed an abnormal peak (31.2%) at the retention time of 4.49 min, which was the position of the S-window and a high value of Hb A2 (4.5%) (A). Compared with HPLC, another method was performed using CE, and the result showed an abnormal peak accounting for 31.8% of the total Hb in zone 7 and Hb A2 3.7% (B). To the best of our knowledge, the most frequent Hb sub-type found in this position is Hb F. In other words, the Hb variant was not completely separated from Hb F, so it can easily be mistaken for Hb F. HPLC results of his wife presented an abnormal peak co-eluted with Hb A2 (A). We collected her cord blood for testing after their baby was born a few months later. The Hb analysis results of the baby were shown in B, and no abnormalities were found. Since the chromatogram of cord blood detected by HPLC is not easy to distinguish, our laboratory does not use HPLC to screen for thalassemia in cord blood.
Figure 1. Hb analysis of the proband with Hb Jiangnan by HPLC and CE. HPLC showed an abnormal peak at retention time 4.49 min(A). CE demonstrated a high Hb X level (31.8%) in the Hb F Zone which were actually the sum of Hb X + Hb F(B).
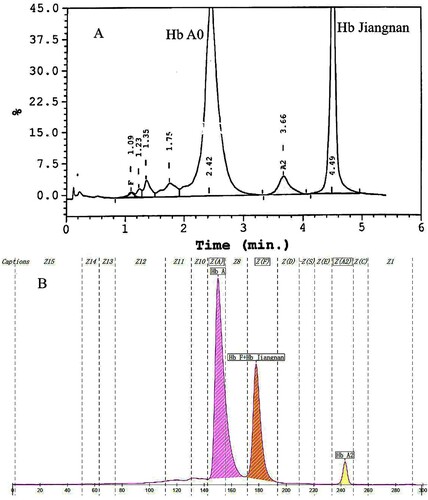
Figure 2. Hb analysis of the proband's wife using HPLC and his baby using CE. HPLC presented a Hb E co-eluted with Hb A2 at retention time 3.67 min in the proband’s wife (A). CE revealed normal results in his baby and there should be a small amount of Hb Jiangnan and Hb F that failed to separate (B).
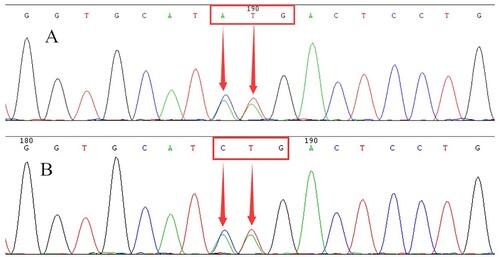
In order to exclude the presence of thalassemia, we conducted a common thalassemia gene analysis. No mutations were detected from the proband, but the CD26 (Hb E) heterozygosity in the β-globin gene was detected from his wife using PCR and reverse dot blot (). DNA sequencing of the β-globin gene identified a heterozygous mutation at position codon 3, resulting in the replacement of the leucine by a lysine residue [β3 (NA3) Leu→Lys, HBB:c.10_11delinsAA] (). This mutation has not been reported in any previous study to the best of our knowledge. DNA sequencing of the baby also suggested that she had inherited the mutation from his father a few moths later.
Figure 3. The results of the proband's wife showed the Hb E heterozygotes by PCR-RDB. No mutations were detected in the proband (A) but a Hb E heterozygote (arrow) was detected from the proband's wife (B).

Figure 4. DNA sequencing of the proband (A) and his baby (B). They revealed a heterozygous CTG>AAG mutation at codon 3 of the β-globin gene (arrow).
Discussion
Hb variants have minimal or no symptoms, so CBC often does not detect them [9,10]. They can be detected through Hb sub-types analysis. Conventional diagnostic tools such as CE and HPLC can be sufficient to detect most Hb variants. However, the detection efficiency of these two methods is not the same [11]. As we reported previously, Hb New York was detected by CE but HPLC [8]. In this study, HPLC revealed that the Hb variant could be separated as an S-window from Hb A0, Hb F, and Hb A2, but CE presented a high-level peak in the overlap of Hb F and Hb variant. In other words, Hb F and Hb variants cannot be separated with CE. If there was no indication of HPLC results, we might consider it an elevated Hb F on the CE program.
For a high level of Hb F cases, hereditary persistence of fetal hemoglobin (HPFH) is suspected firstly after we rule out the possibility of β-thalassemia intermedia or β-thalassemia major [12]. Gap-PCR will be used to detect the deletions of HPFH, and point mutations of HPFH will be detected by DNA sequencing according to our routine identification strategy. Obviously, we can not obtain a correct result from this case if HPFH was done. Therefore, this suggests a suspected high level of Hb F in the CE detection results, and HPLC should be recommended to do a comparison test.
As seen in , HPLC displayed an unknown Hb variant in the S-window. We were able to infer the Hb variant that could be a mutation of the β-globin chain based on the value of the Hb variant (Hb X 31.2%). So, a β-globin gene was amplified and sequenced with our previous report's conditions. The DNA sequencing’s result revealed a mutation (CTG>AAG) at codon 3 in the β-globin gene of the proband. After searching Pubmed and consulting the Hb Var and Ithanet databases, it is the first Hb variant reported at this position. Therefore, we named this novel variant Hb Jiangnan based on the geographic origin of the proband. Subsequently, we successfully obtained the registration from the Ithanet databases (No.3794).
Interestingly, Hb A2 level measured using HPLC was 4.5%, suggesting that the patient was suspected of β-thalassemia. However, neither routine thalassemia genetic analysis nor DNA sequencing supported the existence of β-thalassemia. Similarly, the CE results also showed an elevated Hb A2 (3.7%), but the value was lower than HPLC. Actually, we can also find this situation from Hb E, which can cause a high level of Hb A2. In order to confirm our suspicion, his parents were advised to take blood samples for testing, but they did not follow our suggestion. This high Hb A2 level observation confused the couple's prenatal diagnosis because his wife was a carrier of Hb E. As far as we know, a patient with a β-thalassemia trait combined with Hb E will be β-thalassemia intermedia [13]. Based on normal hematological data, it was likely the glycated fraction (or another adduct) could elute at the position of normal Hb A2, leading to elevated Hb A2. Finally, the possibility of genetic counseling and the prenatal diagnosis was not provided to the couple, but they were told to pay attention to follow-up.
Other mutations described at this location are Hb Kamakura [β3(NA3) Leu→Val, HBB:c.10C>G], Hb Niguarda [β3(NA3) Leu→Met, HBB:c.10C>A], Hb Jabalpur [β3(NA3) Leu→Pro, HBB:c.11T>C], Hb Sedgwick [β3(NA3) Leu→Arg, HBB:c.11T>G], and Hb Santo Domingo [β3(NA3) Leu→Gln, HBB:c.11T>A]. All of them have been reported to be clinically silent and have no high level of Hb A2. Isopropanol stability testing of the proband was negative, and the patient’s red cell indices were normal, suggesting that Hb Jiangnan is also a clinically benign variant. However, Pathogenicity prediction programs Mutation Taster displayed that Hb Jiangnan may be deleterious. Of course, the results of these predicted programs are often known to be unreliable.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Sabath DE. Molecular diagnosis of thalassemias and hemoglobinopathies: an ACLPS critical review. Am J Clin Pathol. 2017;148 (1):6–15.
- Greene DN, Vaughn CP, Crews BO, et al. Advances in detection of hemoglobinopathies. Clin Chim Acta. 2015;439:50–57.
- Zhao YL, Lin QF, He XW, et al. Hb Hezhou [β64(E8)Gly→Ser; HBB: c.193G>A]: a novel variant on the β-globin gene. Hemoglobin. 2021;45 (2):133–135.
- Xu AP, Chen WD, Xie WJ, et al. Identification of a new hemoglobin variant Hb Liuzhou [HBA1:C.182A→G] by MALDI-TOF mass spectrometry during HbA1c measurement. Scand J Clin Lab Invest. 2020;80 (6):479–483.
- Giordano PC, Cnossen MH, Joosten AMS, et al. Codon 24 (TAT>TAG) and codon 32 (ATG>AGG) (Hb Rotterdam): two novel alpha2 gene mutations associated with mild alpha-thalassemia found in the same family after newborn screening. Hemoglobin. 2010;34(4):354–365.
- Baruah A, Baruah MK. Phenotypic diversity and clinico-hematological profile of Hb E-Beta thalassemic children. Indian J hematol blood transfuse. 2020;36 (1):117–122.
- Chuansumrit A, Sirachainan N, Kitpoka P, et al. The Effect of blood transfusion on growth of patients with Hb E/β-Thalassemia. Hemoglobin. 2019;43 (4-5):264–272.
- Li YQ, Huang HP, Chen ZZ, et al. Comparison of capillary electrophoresis and high performance liquid chromatography for detection and quantification of hemoglobin New York. Clin Chem Lab Med. 2016;54 (1):91–95.
- Wang Y, Zhang K, Xu ZY, et al. The presence of Hb J-Bangkok caused spuriously low glycated hemoglobin value on the Tosoh G7. Scand J Clin Lab Invest. 2014;74 (8):725–727.
- Li YQ, Li YW, Liang L, et al. First detection of Hb Cenxi [β46(CD5)Gly→Arg (G GG> C GG), HBB: c.139G>C] by capillary electrophoresis. Hemoglobin. 2021;45 (4):262–264.
- Higgins T, Mack M, Khajuria A. Comparison of two methods for the quantification and identification of hemoglobin variants. Clin Biochem. 2009;42 (7-8):701–705.
- Hariharan P, Kishnani P, Sawant P, et al. Genotypic-phenotypic heterogeneity of δβ-thalassemia and hereditary persistence of fetal hemoglobin (HPFH) in India. Ann Hematol. 2020;99 (7):1475–1483.
- Bohara VV, Ray S, Chakrabarti P, et al. Optimizing the dose of hydroxyurea therapy for patients with β-thalassemia intermedia (Hb E-β-thalassemia): a single center study from Eastern India. Hemoglobin. 2014;38(1):44–48.