
ABSTRACT
Objective
Calreticulin (CALR) mutations have been identified as driver mutations in a quarter of patients with essential thrombocythaemia (ET) and primary myelofibrosis (PMF), which are subgroups of myeloproliferative neoplasms (MPNs). A 52-bp deletion (type I mutation) and a 5-bp insertion (type II mutation) are the most frequent variants. To better understand the impact of different CALR mutant variants, with or without nondriver mutations, on the clinical subtypes of MPN needs further investigation.
Methods
The clinical characteristics, laboratory parameters and genetic mutation statuses were analysed in a cohort of 77 MPN patients with CALR mutations (ET = 24, prePMF = 33, and overt PMF = 20). Targeted NGS using a 38-gene panel was performed to evaluate the variant allele frequency (VAF) of CALR type I/type II mutations and assess the molecular landscape of nondriver gene mutations.
Results
A lower VAF of type I vs. type II was observed in CALR-mutant ET, prePMF and overt PMF, and a higher frequency of type I vs. type II was found in CALR-mutant overt PMF. Additional somatic mutations were indicated to be useful in understanding the pathogenesis of MPN. In this study, the mutation landscape was more complex in overt PMF than in ET or in prePMF. Mutations in epigenetic regulators (ASXL1, EZH2 and TET2) were more common in overt PMF.
Conclusions
The two different subtypes of CALR mutations may have different impacts on MPN. A lower VAF of CALR type I indicates a greater contribution to disease progression in MPN, and increased nondriver mutations may be important in myelofibrosis progression.
Introduction
Somatic mutations of calreticulin (CALR) were identified as driver mutations of myeloproliferative neoplasms (MPNs) in 2013, which were found in JAK2/MPL-unmutated essential thrombocythemia (ET) and primary myelofibrosis (PMF), three subsets of MPNs. More than 50 different types of CALR mutations have been identified. These mutations are +1 frameshift mutations caused by a small deletion or insertion of nucleotides in the reading frame of exon 9, which is the terminal exon of CALR. The most common CALR variants are a 52-bp deletion (type I mutation) and a 5-bp insertion (type II mutation) [Citation1, Citation2].
As a context-dependent oncogene, mutant CALR is associated with the thrombopoietin (TPO) receptor MPL and constitutively activates JAK/STAT signalling. Thus, it contributes to the pathogenesis of ET and PMF [Citation3–5]. Recent studies have found that in retroviral mouse modelling, CALRdel52 (type I mutant)-expressing mice could develop ET-like changes and progress to myelofibrosis over time, while CALRins5 (type II mutant)-expressing mice only had ET-like changes without myelofibrosis [Citation6, Citation7]. Moreover, two previous studies found a significantly higher risk of fibrotic progression in ET patients with type I / I -like CALR mutations than in those with type II / II -like CALR mutations, and the increased CALR allele burden was associated with disease progression [Citation8, Citation9]. Nevertheless, the pathological mechanism of how different subtypes of CALR mutations affect the occurrence and progression of MPN is not fully understood, and further studies are needed to confirm.
In recent years, additional MPN-related somatic mutations were identified to influence thrombosis and disease progression in MPN [Citation10, Citation11]. The most commonly confirmed mutations were in genes involved in epigenetic regulation, cytokine signalling pathways, spliceosome regulators, and transcription factors. These mutations were not disease specific but were recently included in a diagnostic algorithm [Citation12].
With the further understanding of MPN, prefibrotic/early PMF (prePMF) was included in the 2016 WHO classification of myeloid neoplasms as a subtype of PMF for the first time, and it had a different clinical picture and outcomes from ET [Citation13, Citation14]. Due to the heterogeneity of prePMF, there is no universally applicable treatment strategy for prePMF. In this study, we investigated the clinical features and the relationship between the distinct mutation landscape and clinical phenotype in CALR-mutant ET, prePMF and overt PMF.
Materials and methods
Study population
Our cohort included 77 consecutive patients with CALR mutations diagnosed with ET, prePMF or overt PMF according to the 2016 WHO criteria at the Second Hospital of Hebei Medical University, Shijiazhuang, between 2017 and 2020[Citation13]. Bone marrow samples were obtained after they provided written informed consent and after ethics approval from relevant authorities was obtained.
All the clinical and molecular variables analyzed in the study were collected from newly diagnosed patients, including the variant allele frequency (VAF) of CALR mutations and bone marrow pathology. The prognostic model, including International Prognostic Score for Essential Thrombocythemia (IPSET)-survival score for ET patients, MIPSS70 score for prePMF and overt PMF patients, and IPSET-thrombosis score for ET and prePMF patients, were as previously described [Citation15–17]. ().
Table 1. Clinical and laboratory characteristics of 25 ET patients stratified according to the CALR mutant subtypes.
Table 2. Clinical and laboratory characteristics of 32 prePMF patients stratified according to the CALR mutant subtypes.
Table 3. Clinical and laboratory characteristics of 20 overt PMF patients stratified according to the CALR mutant subtypes.
CALR and nondriver mutation analysis
Targeted NGS using the 38-gene panel (Yuanqi, Shanghai, China) was performed to determine the CALR mutation types and whether there were other nondriver genetic mutations. Genomic DNA was extracted from bone marrow samples using QIAamp DNA mini kit (Qiagen, Valencia, CA, USA). Genomic DNA quality and quantity were determined using a Nanodrop 8000 UV-Vis spectrometer (Thermo Scientific, Waltham, MA, USA), a Qubit 2.0 Fluorometer (Life Technologies Inc., Grand Island, NY, USA), and a 2200 TapeStation Instrument (Agilent Technologies, Santa Clara, CA, USA). As previously reported, this panel is designed to enrich exons of 38 genes covering 66.9 kb of the human genome (including CALR, JAK2, MPL, ASXL1, EZH2, SRSF2, IDH1, IDH2, U2AF1, TET2, STAG2, BCOR, BCORL1, DNMT3A, CBL, CEBPA, MLL, SF3B1, ATM, GATA2, NRAS, KRAS, RUNX1, PPM1D, ZRSR2, KIT, SETBP1, NF1, TP53, NPM1, PDGFRA, PHF6, PIGA, SH2B3, FLT3, STAT3, CSF3R, WT1). After the enriched exome libraries were multiplexed, the libraries were sequenced using the NovaSeq 6000 sequencing platform (Illumina, San Diego, CA, USA).
Statistical analysis
Numerical variables are shown by their median and range, and categorical variables are shown by count and relative frequency (%). Comparisons of quantitative variables between two groups of patients were performed by the nonparametric Wilcoxon rank-sum test. Associations between categorical variables (two-way tables) were tested by Fisher’s exact test.
Statistical analyses were performed using SPSS 19.0 software, and P values <0.05 were considered statistically significant.
Results
Characteristics of MPN patients with CALR type I/type II mutations
In total, 46 patients carried CALR type I mutations, including 13 patients with ET (5 males, 8 females; median age: 66; age range: 28–69 years), 18 patients with prePMF (10 males, 8 females; median age: 60; age range: 18–76 years) and 15 patients with overt PMF (8 males, 7 females; median age: 63; age range: 42–74 years). 31 patients carried CALR type II mutations, including 12 patients with ET (3 males, 9 females; median age: 55; age range: 25–78 years), 14 patients with prePMF (4 males, 1 female; median age: 64; age range: 27–89 years) and 5 patients with overt PMF (5 males, 8 females; median age: 64; age range: 56–74 years). Their clinical characteristics are summarized in . Comparing the patients carrying CALR type I and type II mutations, the age, sex, white blood cell counts, platelet counts, haemoglobin, serum LDH, the incidence of splenomegaly, the grade of fibrosis and different risk distributions were not significantly different in ET, prePMF and overt PMF (P > 0.05) ().
Mutation load and incidence of CALR type I/type II mutations in ET, prePMF and overt PMF
The VAF of CALR type I mutations was significantly lower than that of CALR type II mutations in ET (19.38% vs. 40.51%, P < 0.001), prePMF (18.61% vs. 45.24%, P < 0.001) and overt PMF (19.11% vs. 44.88%, P < 0.001) ().
Figure 1. The VAF of CALR type I/type II found in 77 patients with ET, prePMF and overt PMF.
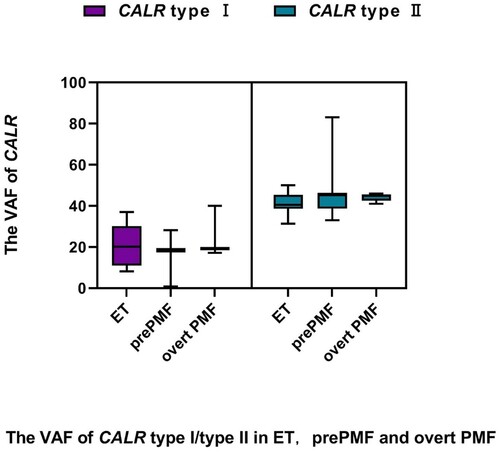
The frequency of CALR type I mutations was significantly higher than that of CALR type II mutations in overt PMF (75% vs. 25%, P = 0.025). However, there were no significant differences in the frequency between the two CALR variants in ET (52% vs. 48%, P = 0.841) or in prePMF (56% vs. 44%, P = 0.480).
Nondriver mutation status in MPN patients with CALR type I/type II mutations
Compared with ET and prePMF, nondriver mutations were more frequently mutated in overt PMF patients. (overt PMF 70% vs. ET 24%, P = 0.002; overt PMF 70% vs. prePMF 25%, P = 0.001). Furthermore, in the 14 overt PMF patients with additional mutations, 8 patients had more than three nondriver mutations, and the other 6 patients had one or two nondriver mutations with a heavy mutation load (VAF >30%) (b).
Figure 2. Mutation patterns. (a) Numbers of nondriver mutations in the respective genes are shown. Colour code represents myeloproliferative neoplasm (MPN) subgroups as indicated. (b) Distribution of mutated genes is shown on a single sample level. Each column represents one patient sample. Patients are divided into subgroups of MPN with CALR type I/type II mutation. The shades of red represent the mutant load of the gene as indicated.
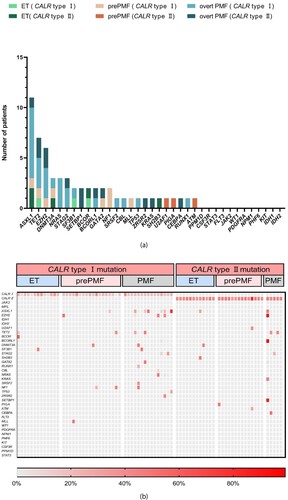
Comparing the two groups of patients with CALR type I and type II mutations, there was no significant difference in the frequency of nondriver genes in ET, prePMF and overt PMF patients (ET: 15.4% vs. 25.0%, P = 0.645, prePMF: 33.3% vs. 14.3%, P = 0.412, overt PMF: 66.7% vs. 80.0%, P = 1.000) ().
Among these 77 patients, mutations in epigenetic regulators (ASXL1, EZH2 and TET2) were more frequent in those with overt PMF. The most frequently mutated gene was ASXL1, with 11 mutations. Of these, 8 were in overt PMF patients (CALR type I mutation = 7, CALR type II mutation = 1), including one patient with two concurrent variants. Additionally, 4/5 EZH2 mutations (CALR type I mutation = 3, CALR type II mutation = 1), and 5/7 TET2 mutations (CALR type I mutation = 3, CALR type II mutation = 2) were detected in overt PMF patients (a).
Twenty genes, namely, DNMT3A (n = 3), BCOR (n = 3), STAG2 (n = 3), NRAS (n = 3), SF3B1 (n = 2), GATA2 (n = 2), NF1 (n = 2), SETBP1 (n = 2), SRSF2 (n = 1), U2AF1 (n = 1), CBL (n = 1), KRAS (n = 1), ZRSR2 (n = 1), SH2B3 (n = 1), TP53 (n = 1), MLL (n = 1), RUNX1 (n = 1), PIGA (n = 1), CEBPA (n = 1) and ATM (n = 1), were mutated in less than 5% of all 77 patients (b).
Discussion
In this study, we analysed 77 patients with CALR-mutant ET, prePMF or overt PMF using a 38-gene NGS panel to evaluate their clinical phenotype and molecular landscape. We found that the VAF of CALR type I mutations was lower than that of CALR type II mutations in all three subgroups. Previous studies have described that CALR mutations in MPN patients accumulate in a narrow region of exon 9, resulting in a gain-of-function mutation [Citation1, Citation2]. All the CALR variants share amino acid sequences at their carboxyl(C)-terminal end, which replaces a cluster of negatively charged amino acids at the C-terminal end of wild-type (wt) CALR. CALR type I mutations contain a more mutant-specific domain than CALR type II mutations, which create mutant proteins with highly altered calcium binding activity [Citation18]. The structural diversity of different subtypes of CALR mutations may have different impacts on MPN. We speculate that CALR type I mutations maybe cause various phenotypes of MPN at low mutation load.
In addition, in the three subgroups of MPN, we also found that type I mutations were significantly more frequent than CALR type II mutations in overt PMF patients, but not in ET and prePMF patients, that was consistent with the findings of previous studies. [Citation18, Citation19]. Phenotypic differences between the two CALR mutant subtypes were also consistently observed between type I-like mutations, such as del19, and type II-like mutations, such as del34 and del36[Citation20]. The study by Ilyas Chachoua’s group showed that in contrast to CALR type II mutations, CALR type I mutations provided a greater growth advantage in transduced stem cells, and mice with these mutations were more likely to develop thrombocytosis and the fibrotic phenotype. This might partly explain the higher frequency of type I vs. type II CALR mutations in overt PMF [Citation3]. In our study, although lower VAF of CALR type I mutations was found in each subgroup of MPN patients, CALR type I mutations were more common in patients with overt PMF, suggesting that CALR type I mutations may be more likely to contribute to disease progression in MPN. Further follow-up of this group of patients is necessary to confirm this conclusion.
With increasing knowledge of the molecular basis of MPN, it is becoming evident that both driver and nondriver mutations have relevance in disease progression or transformation [Citation21, Citation22]. In our study, nondriver mutations were more prevalent in overt PMF. Some overt PMF patients had more than 3 nondriver mutations, and the other patients had a heavy mutation load. These results suggest that patients with overt PMF have a highly complex molecular landscape. Mutations in specific genes, such as the high molecular risk mutations of ASXL1 and EZH2, were the most common nondriver mutations in overt PMF patients, especially in those carrying CALR type I mutations [Citation20, Citation23]. Recent research has shown that epigenetic regulators have a prognostic impact on overt PMF and may modify treatment strategies. However, type I-like CALR mutations in overt PMF may partially amend the detrimental effect of high molecular risk mutations, such as ASXL1[Citation21].
As a prodromal phase of PMF, prePMF has been recognized as an independent subtype in the 2016 WHO classification of myeloid neoplasms. It is different from true ET, in particular the higher rates of progression to overt myelofibrosis and blast transformation [Citation13, Citation24–27]. Furthermore, it has become increasingly clear over recent years that nondriver mutations have an impact on disease progression or transformation [Citation25]. In our study, we found the number of nondriver mutations was higher in type I vs. type II CALR-mutated prePMF patients, but there was no statistical difference between the two groups. As a heterogeneous disease, prePMF ranges from isolated thrombocytosis, mimicking essential thrombocythemia (ET), to symptoms of high-risk PMF, and it may be more appropriate to analyze nondriver mutations in prePMF patients after risk stratification to gain a more accurate understanding of prePMF. Unfortunately, the sample size of this study was limited, and larger samples are needed for further research.
In conclusion, molecular testing in MPN, including driver and nondriver mutations, has proven to be useful for advancing the understanding of the heterogeneity of this disease. Our results suggest that the two CALR variants may have different effects on the pathogenesis of MPN. In CALR-mutant overt PMF patients, the number and the different VAFs of nondriver genes make the molecular landscape more complex, which might have an impact on the course and outcome of the disease. Further follow-up of this group of patients and larger clinical studies are needed to confirm the value of the complex molecular status to better understand the dynamics of MPN.
Disclosure statement
No potential conflict of interest was reported by the author(s).
References
- Klampfl T, Gisslinger H, Harutyunyan AS, et al. Somatic mutations of calreticulin in myeloproliferative neoplasms. N Engl J Med. 2013;369(25):2379–2390.
- Nangalia J, Massie CE, Baxter EJ, et al. Somatic CALR mutations in myeloproliferative neoplasms with nonmutated JAK2. N Engl J Med. 2013;369(25):2391–2405.
- Chachoua I, Pecquet C, El-Khoury M, et al. Thrombopoietin receptor activation by myeloproliferative neoplasm associated calreticulin mutants. Blood. 2016;127(10):1325–1335.
- Pecquet C, Chachoua I, Roy A, et al. Calreticulin mutants as oncogenic rogue chaperones for TpoR and traffic-defective pathogenic TpoR mutants. Blood. 2019;133(25):2669–2681.
- Nami M, Marito A, Yinjie Y, et al. Mutant calreticulin interacts with MPL in the secretion pathway for activation on the cell surface. Leukemia. 2020;34(2):499–509.
- Marty C, Pecquet C, Nivarthi H, et al. Calreticulin mutants in mice induce an MPL-dependent thrombocytosis with frequent progression to myelofibrosis. Blood. 2016;127(10):1317–1324.
- Li J, Prins D, Park HJ, et al. Mutant calreticulin knockin mice develop thrombocytosis and myelofibrosis without a stem cell self-renewal advantage. Blood. 2018;131(6):649–661.
- Bertozzi I, Biagetti G, Vezzaro T, et al. Clinical effect of CALR allele burden in patients with essential thrombocythemia. Ann Hematol. 2022;101(6):1345–1346.
- Loscocco GG, Guglielmelli P, Gangat N, et al. Clinical and molecular prdictors of fibrotic progression in essential thrombocythemia: A multicenter study involving 1607 patients. Am J Hematol. 2021;96(11):1472–1480.
- Tefferi A, Guglielmelli P, Lasho TL, et al. Mutation-enhanced international prognostic systems for essential thrombocythaemia and polycythaemia vera. Br J Haematol. 2020;189(2):291–302.
- Guglielmelli P, Gangat N, Coltro G, et al. Mutations and thrombosis in essential thrombocythemia. Blood Cancer J. 2021;11(4):77.
- Grinfeld J, Nangalia J, Baxter EJ, et al. Classification and personalized prognosis in myeloproliferative neoplasms. N Engl J Med. 2018;379(15):1416–1430.
- Barbui T, Thiele J, Gisslinger H, et al. The 2016 WHO classification and diagnostic criteria for myeloproliferative neoplasms: document summary and in-depth discussion. Blood Cancer J; 2018;8(2):15.
- Barosi G, Rosti V, Bonetti E, et al. Evidence that prefibrotic myelofibrosis is aligned along a clinical and biological continuum featuring primary myelofibrosis. PLoS ONE. 2012;7(4):e35631.
- Passamonti F, Thiele J, Girodon F, et al. A prognostic model to predict survival in 867 World Health Organization-defined essential thrombocythemia at diagnosis: a study by the International Working Group on Myelofibrosis Research and Treatment. Blood. 2012;120(6):1197–1201. (IPSET-survival).
- Barbui T, Finazzi G, Carobbio A, et al. Development and validation of an International Prognostic Score of thrombosis in World Health Organization-essential thrombocythemia (IPSET-thrombosis). Blood. 2012;120(26):5128–5133.
- Guglielmelli L, Lasho TL, Rotunno G, et al. MIPSS70: Mutation-Enhanced International Prognostic Score System for Transplantation-Age Patients With Primary Myelofibrosis. J Clin Oncol. 2018;36(4):310–318.
- Pietra D, Rumi E, Ferretti VV, et al. Differential clinical effects of different mutation subtypes in CALR-mutant myeloproliferative neoplasms. Leukemia. 2016;30(2):431–438.
- Cabagnols X, Defour JP, Ugo V, et al. Differential association of calreticulin type 1 and type 2 mutations with myelofibrosis and essential thrombocytemia: relevance for disease evolution. Leukemia. 2015;29(1):249–252.
- Agarwal R, Blombery P, McBean M, et al. Clinicopathological differences exist between CALR- and JAK2-mutated myeloproliferative neoplasms despite a similar molecular landscape: data from targeted next-generation sequencing in the diagnostic laboratory. Ann Hematol. 2017;96(5):725–732.
- Tefferi A, Guglielmelli P, Lasho TL, et al. CALR and ASXL1 mutations-based molecular prognostication in primary myelofibrosis: An international study of 570 patients. Leukemia. 2014;28:1494–1500.
- Tefferi A, Nicolosi M, Mudireddy M, et al. Driver mutations and prognosis in primary myelofibrosis: Mayo-Careggi MPN alliance study of 1,095 patients. Am J Hematol. 2018;93(3):348–355.
- Cottin L, Riou J, Orvain C, et al. Sequential mutational evaluation of CALR -mutated myeloproliferative neoplasms with thrombocytosis reveals an association between CALR allele burden evolution and disease progression. Br J Haematol. 2020;188(6):935–944.
- Gisslinger H. Pre-PMF emerging as important subgroup of MPN. Blood. 2017;129(24):3142–3144.
- Guglielmelli P, Pacilli A, Rotunno G, et al. Presentation and outcome of patients with 2016 WHO diagnosis of prefibrotic and overt primary myelofibrosis. Blood. 2017;129(24):3227–3236.
- Jeryczynski G, Thiele J, Gisslinger B, et al. Pre-fibrotic/early primary myelofibrosis vs. WHO-defined essential thrombocythemia: the impact of minor clinical diagnostic criteria on the outcome of the disease. Am J Hematol. 2017;92(9):885–891.
- Carobbio A, Guglielmelli P, Rumi E, et al. A multistate model of survival prediction and event monitoring in prefibrotic myelofibrosis. Blood Cancer J. 2020;10(10):100.