
ABSTRACT
Aim
To evaluate the contributions of VWF to the clinical manifestation and severity of SCD
Design
A systematic review of peer-reviewed articles published in English. The review was carried out in accordance with the Preferred Reporting Items for Systematic Reviews and Meta-Analyses checklist.
Methods
The data sources for the review included MEDLINE, PubMed, CINAHL, and Academic Search Complete. Articles that applied a quantitative approach to the investigation of the relationship of vWF with clinical manifestations and severity indices were included. The risk of bias assessment was carried out with a mixed-method appraisal tool. We computed I 2 to estimate the degree of heterogeneity.
Result
There was a significantly higher level of VWF in SCD than in the control (d = 2.7, Z = 4.865, P < 0.001, I 2 = 96.41%). Significant positive correlations were obtained for the relationship of VWF with vasoocclusive crisis (r= 0.277, Z= 5.077, P < 0.001, 1 2 =15.62), rate of hemolysis (r=0.441; Z= 4.440, I 2 = <1%), extracellular haemoglobin (r=-0.397, Z=-4.155, I 2 =<1%) and CRP (r = 0.331, Z = 4.566, P < 0.001, I 2 < 1%).
The VWF is important in determining the clinical severity of sickle cell disease, which constitutes a putative therapeutic target. More work is required to understand the causal direction underlying the association between VWF levels and the clinical severity of sickle cell disease and the potential role that VWF plays in the clinical manifestations of sickle cell disease.
Protocol registration
The protocol was registered with PROSPERO (CRD42021262625).
Introduction
Sickle cell disease (SCD) is a group of autosomal recessive disorders characterized by phenotypic variations [Citation1]. Two copies of a mutant -globin allele or an allele that specifies a deficient or faulty -globin allele cause SCD [Citation2]. As a result, erythrocytes are rigid with increased expression of adhesion molecules. They adhere and occlude vessels, leading to ischemia/reperfusion injury with oxidative stress and damage-induced cell lysis [Citation3]. These pathogenic mechanisms explain clinical symptoms such as chronic hemolytic anemia seen in sickle cell anemia, a subtype of SCD, and episodic vaso-occlusive crisis. Individuals with elevated hemolytic rates run the risk of developing a syndrome characterized by leg ulcers, pulmonary hypertension, priapism, and its resultant side effect of sexual dysfunction [Citation4].
Activation of the coagulation system plays a role in the pathogenesis of SCD. SCD is considered a prothrombotic condition associated with chronic activation of the coagulation system. This is associated with increased plasma levels of prothrombin fragment 1,2 and thrombin-antithrombin complexes, which are markers of thrombin generation [4b]. There is evidence of increased platelet activation and expression of adhesion molecules that mediate binding to more fibrinogen (αiibβ-) SCD is also associated with elevated plasma levels of von Willebrand factor (VWF) [Citation5].
VWF is a glycoprotein that is known to mediate platelet adhesion, increased platelet adhesion and aggregation, and impaired fibrinolytic activity, all of which contribute to a state of hypercoagulable and prothrombotic disease [Citation5]. VWF is a large sticky multimeric protein that contributes to vascular hemostasis and functions as a marker of endothelial dysfunction [Citation6]. The larger the protein, the more adhesive it is to function. A VWF deficiency or defect can result in von Willebrand disease and bleeding. On the other hand, an increase in VWF can promote thrombosis by creating a favourable environment [Citation7]. Hypercoagulability has been linked to a variety of clinical symptoms, including severe vasoocclusive crises, hemolytic anemia, and oxidative stress [Citation3,Citation5]. In SCD, a similar relationship has been discovered between an elevated level of extracellular hemoglobin (Hb) and the development of the disease. Extracellular Hb binds to VWF and inhibits its cleavage by ADAMTS-13, resulting in the accumulation of ultra-large VWF multimers in the circulation and in the endothelium, as well as the stimulation of prothrombotic events [Citation8].
However, the cumulative effect of VWF on clinical indices relevant to people with SCD remains unknown. For instance, Akaba et al. [Citation5] reported no correlation between VWF and hematocrit; Omer et al. [Citation9] discovered a correlation between VWF level and severity score; Chen et al. [Citation10] reported a correlation between hemolysis rate and the amount of active VWF in plasma [Citation10], and Ahmed et al. [Citation11] found no correlation between VVWF and hematocrit (hemolysis). Additionally, the extent to which VWF can be used to predict disease severity remains unknown in studies that found a positive correlation between VWF and SCD severity. Despite this, it is currently believed that the Hb-VWF interaction is a therapeutic target for the treatment of thrombotic and vasoocclusive complications in patients with SCD with severe intravascular hemolysis, making VWF the most important adhesive molecule/target in the pursuit of good clinical outcomes in people with SCD [Citation5,Citation8,Citation12]. Surprisingly, despite the methodical resolution of scholarly disagreements, the cumulative contribution to the clinical symptoms or severity of SCD has not been clarified [Citation6]. As a result, the objective of this systematic review is to determine the effect of VWF on the clinical manifestations of SCD in terms of severity score, vaso-occlusion, hemolysis rate, thromboembolic events, and hematological indicators.
Aim
The purpose of this study is to determine the role of the von Willebrand factor in the clinical manifestations and severity of sickle cell disease.
Methodology
Design
This is a systematic review of cross-sectional and longitudinal observational studies. The protocol was structured according to the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) standard [Citation13]. The protocol was registered with PROSPERO (CRD42021262625).
Eligibility criteria
Study characteristics
This review involved peer-reviewed articles published in English regardless of location, sample size, or test statistics.
Participants: Individuals diagnosed with SCD. Studies were included regardless of whether or not hematological confounding factors were assessed.
Intervention: Not applicable. This is a systematic review of observational studies that investigated the association between VWF and clinical manifestations and/or severity of SCD.
Control: studies were included regardless of whether or not a control group was used to study the subject.
Inclusion criteria
Peer-reviewed articles that described the relationship between VWF and one or more of the following variables: vascular occlusive crisis, thromboembolic event, hemolysis rate, overall severity, hematocrit, hypoxia, and platelet counts.
English-language peer-reviewed articles
Exclusion criteria
The grey literature was excluded
Publications in languages other than English.
Studies in which outcome variables were estimated using substandard techniques.
Sources of information and search strategy
PubMed, MEDLINE, CINAHL and Academic Search Complete were searched using medical subject headings (MeSH) and keywords identified in the title, abstract, and/or text of the articles. PubMed was used to test the search strategy. The MeSH terms identified through the Cochrane MeSH terms were included in the pilot search. After experimenting with various combinations of these terms, the most sensitive strategy was selected and documented. Sensitivity judgments were made face-to-face. The strategy was modified to accommodate the remaining databases’ syntax and subject headings (MEDLINE, CINAHL, and Academic Search Complete).
Management of records and data
Results were exported directly from databases into EndNote 8, where deduplication and initial screening were carried out. Following an initial screening, the bibliographic records were exported to COVIDENCE (Extraction 2) for independent review by two independent reviewers. To help the screening process, we used a pilot and refined screening template that includes eligibility questions.
Data selection
An expert reviewer conducted an initial screening of the title and abstract to identify those that met the inclusion criteria. Subsequently, two reviewers conducted an independent screening of the study titles and abstracts. In consultation with the expert reviewer, any conflict was resolved. Any disagreements were resolved until the team agreed on what articles to include. A PRISMA diagram was used to illustrate the flow of studies throughout the selection process, as well as the reasons for exclusion ().
Figure 1. PRISMA Flow Diagram of the contributors of von Willebrand factor to clinical severity of sickle cell disease: a systematic review (1998-2020).
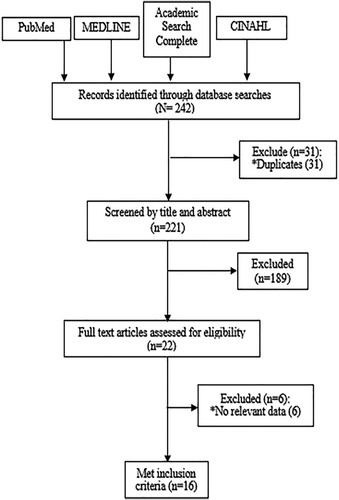
Data collection process.
Quality evaluation and risk of bias assessment
The two independent reviewers conducted a quality assessment of the included studies. The mixed method appraisal tool (MMAT) Version 2011 was used to assess quality [Citation14]. Section 4 was used to evaluate descriptive studies. The quality of each study was determined and reported according to the MMAT guidelines [Citation14].
Data items
The primary data sought were the von Willebrand factor level, the vascular occlusive crisis, the rate of hemolysis, global severity, the hematocrit, and other measures of disease severity such as inflammatory markers and hypoxemia/oxygen saturation. Additionally, we obtained from each article the following information: the authors’ names, the study title, the study population, sample size, sampling methods, method of assessment, country, and summary of the findings.
Data synthesis and evaluation of heterogeneity
We used a random-effects meta-analysis to quantify the contribution/relationship between VWF and any or all of the following: vascular occlusive crisis, hemolysis rate, hemoglobin, and inflammatory markers. Data on global severity, hematocrit, and hypoxemia/oxygen saturation were insufficient to warrant a meta-analysis. Measures of heterogeneity, i.e. study characteristics were arranged chronologically and presented on an evidence table (Appendix 2). Following Higgins and Thompson [Citation15,Citation16], we computed the I2 statistic. In this review, I2 was interpreted according to the pattern outlined in the Cochrane Handbook for Systematic Reviews of Interventions: 0-40% heterogeneity is considered low, 30-60% moderate heterogeneity, 50-90% substantial heterogeneity and 75-100% considerable heterogeneity (Cochrane Handbook for Systematic Reviews of Interventions) [Citation17].
Data analysis
The relationship between VWF and each vascular occlusive crisis, rate of hemolysis, hemoglobin and inflammatory markers was quantified through meta-correlation. We used the Egger test to estimate publication bias across studies. The comprehensive meta-analysis software package was used, with the significance level set at 0.05.
Results
Review profile
Following a literature search, we retrieved 242 records. Thirty-one duplicate copies were removed leaving two hundred and twenty-one records for screening. After selecting the title and abstract, we eliminated 189 irrelevant records, leaving 22 for full-text review (). Reasons for elimination during abstract and title selection include the use of the wrong study population, the inappropriate study outcome, and/or lack of a report of the interaction between the von Willebrand factor and the stipulated outcomes of interest. Finally, 16 articles were included in our review, which included 1,099 participants from ten countries, and 14 studies were involved in the meta-analysis. Sample sizes ranged from 13 in Chen et al. [Citation3] to 110 in Akaba et al. [Citation5]. Five (31.3%) of the included studies were conducted in The Netherlands, while two each were conducted in the USA and UK. Most (75%) of the studies were cross-sectional, with the control group employed in 11 (68.8%) of them. The remaining four studies were cohort design. Thirteen (80.1%) studies reported the age of the participants, which averaged 20.4 ± 5.7 years. All studies except one, Akaba et al. [Citation5] omitted information about participants’ level of education of the participants (Appendix 2).
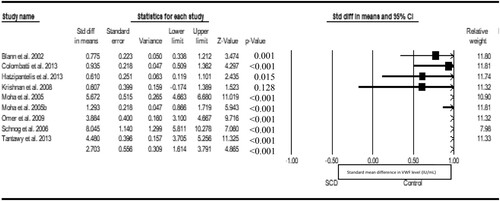
We fitted a random-effect meta-analysis to compare VWF in both SCD patients and non-SCD control. Nine eligible studies were included in the analysis, with the result showing a statistical significantly higher level of VWF in SCD than the control (d = 2.7, Z = 4.865, P < 0.001, I2 = 96.41%), and publication bias (Eggers t value = 3.524, P = 0.00968) ().
Figure 2. Forest plot comparing VWF levels in patients with SCD and control.
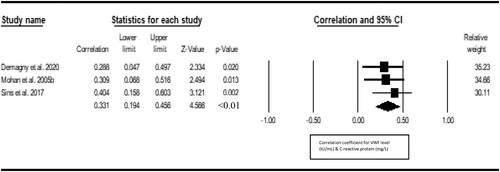
Seven studies were included in the metacorrelation of VWF with vaso-occlusive crisis. The result shows a significant positive correlation between VWF and vasoocclusive crisis (r = 0.277, Z = 5.077, P < 0.001, 12 = 15.62), with publication bias (Eggers t value = 7.273, P = 0.001) (). Three studies were included in the metacorrelation of VWF with the level of hemolysis. We found a statistically significant positive correlation between VWF and hemolysis level of hemolysis (r = 0.441; Z = 4.440, I2 = <1%) and there was no publication bias (Eggers t-value = 1.327, 0.411) (). Three studies were included in the metacorrelation of VWF with fetal hemoglobin (%HbF). The result shows a negative correlation between VWF and %HbF (r = −0.397, Z = −4.155, I2 = <1%), with no publication bias (Egger’s t value = 3.840, p 0.1618) (). Three studies were included in the metacorrelation of VWF with fetal hemoglobin. There was a statistically significant correlation between VWF and C-reactive protein (r = 0.331, Z = 4.566, P < 0.001, I2 < 1%), with associated publication bias (Eggers-t value = 13.182, P = 0.048) (). For insufficient study data, we could not use meta-analysis to examine the relationship of VWF with fibrinogen (2 studies), hypoxia/oxygen saturation (2 studies), interleukin-6 (1 study), haematocrit (1 study) and antioxidant capacity (1 study) and global severity (1 study).
Figure 3. Forest plot showing the correlation between VWF and vasoocclusive crisis.
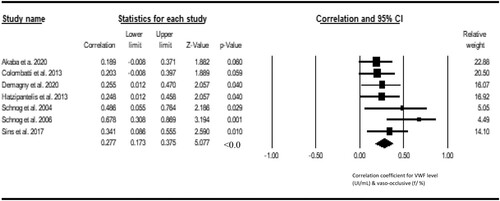
Figure 4. Forest plot showing the correlation between VWF and hemolysis rate.
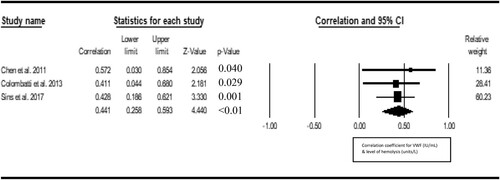
Figure 5. Forest plot showing the correlation between VWF and fetal hemoglobin.
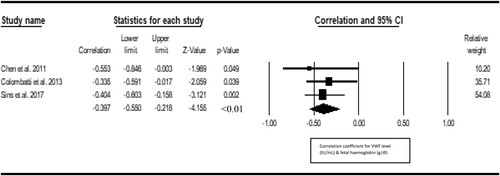
Figure 6. Forest plot showing the correlation between VWF and the C-reactive protein.
However, fibrinogen was correlated (r = 0.317) with VWF in Demagny et al. [Citation18] but not in Omer et al. [Citation9] (r = 0.1, P >0.05). The VWF was correlated with hypoxia (r = 0.557, P = 0.007) and oxygen saturation (r = −0.54, p = 0.01) and global severity of the disease (0.79, P = 0.0085). In two studies [Citation3,Citation5], VWF did not correlate with hemocrit (r = −0.034 & r = 0.05, P > 0.05). Only one study [Citation19] reported the relationship of VWF with antioxidant capacity, with no significant correlation (r = 0.074, p = 0.568).
Discussion
Von Willebrand factor (VWF) is a multimeric glycoprotein that is best known for its ability to mediate platelet adhesion [Citation20]. Acutely activated endothelial cells can release massive amounts of very large and hyper-adhesive VWF molecules capable of spontaneously binding platelets and erythrocytes, particularly sickle cells [Citation21–23]. Normally, VWF is rapidly cleaved by the ADAMTS13 metalloproteinase into smaller, less adhesive multimers, thus avoiding potentially fatal complications such as thrombotic thrombocytopenic purpura [Citation24]. The finding in this review that von Willebrand factor (VWF) levels were higher in SCD patients than in controls without SCD suggests a role for VWF in the pathophysiology and clinical manifestations of SCD. This is consistent with previous findings [Citation9,Citation25–27]; that patients with SCD have elevated levels of hyper-adhesive VWF in their plasma, the amount of which correlates with their hemolysis rate. Differential glycosylation of platelet bound VWF makes it resistant to ADAMTS13 proteolysis [Citation10]. Overexpression of VWF promotes hypercoagulability, which has been associated with a variety of clinical symptoms, including severe vasoocclusive crises, hemolytic anemia, and oxidative stress [Citation3,Citation5].
The positive correlation between VWF and vasoocclusive disease suggests that elevated VWF levels may be associated with an increase in the severity of the disease or clinical manifestations of SCD. This is consistent with previous research [Citation5,Citation9,Citation18], thus reinforcing the postulation that, in steady state, SCD is characterized by higher VWF level in addition to thrombocytosis compared to controls without SCD [Citation9]. There was a positive correlation between VWF and hemolysis, implying that hemolysis in SCD is microangiopathic, with rigid sickle erythrocytes suffering oxidative stress and damage while navigating through a mesh of VWF strands. This notion is supported by the description of a syndrome similar to thrombotic thrombocytopenic purpura in patients with SCD [Citation28,Citation29]. The adhesion of sickle erythrocytes to VWF immobilized with endothelial cells can also increase hemolysis by reducing the transit time of sickled erythrocytes sufficiently to induce hemoglobin deoxygenation and polymerization, which is consistent with the observation that adhesion of sickle erythrocytes to ULVWF was greatest at a fluid shear stress of 0.5 dyne/cm2, which is similar to that of post-capillary venules [Citation30], the site of vaso-occlusion in SCD. Patients with the highest hemolytic rates are at risk of developing a syndrome characterized by pulmonary hypertension, priapism, and leg ulcers; thus, VWF levels could serve as a marker of the severity of the disease in people with SCD.
Similarly, VWF and hemoglobin (Hb) showed a positive correlation. This is consistent with previous findings [Citation3,Citation31,Citation32], suggesting that VWF-induced hemolysis in SCD is mediated by its binding to Hb. A high level of extracellular hemoglobin (Hb) in plasma has been associated with the pathogenesis of SCD [Citation8]. Therefore, patients with SCD are likely to have acquired ADAMTS-13 deficiency, due to Hb competitive binding to and inhibition of VWF proteolysis, resulting in the accumulation of ultra-large VWF multimers in the circulation and endothelium [Citation8]. Hence, the Hb-VWF interaction may be considered a therapeutic target for patients with severe intravascular hemolysis, such as those with SCD, who develop thrombotic and vasoocclusive complications. In summary, increased VWF levels exacerbate the vasocclusive crisis in SCD by increasing hemolysis and extracellular hemoglobin levels in plasma. Furthermore, VWF was also associated with nocturnal hypoxemia and low oxygen saturation [Citation33], although the evidence is scant.
In terms of the interaction between VWF and the inflammatory marker, the findings indicated a positive correlation between VWF and the C-reactive protein. This finding is consistent with the idea that increased VWF reactivity during VOC may be directly related to both inflammation and oxidative stress, both of which are strongly up-regulated during VOC in SCD [Citation32]. Sickle cell anemia (SCA) is characterized by the activation of blood cells and the release of pro-inflammatory molecules such as C-reactive protein, thus perpetuating a chronic inflammatory state that affects blood flow [Citation34]. Individuals who have more vasocclusive crises have a higher chance of experiencing inflammation [Citation35].
The major limitation to this review is the small number of literature involved in the quantitative synthesis. Furthermore, the failure of individual studies to account for the ABO group when examining the putative contribution of VWF to sickle cell manifestations constitutes a limitation. The use of quantitative approach is one of the strengths of the study thus allowing inference to be drawn objectively. Further research is required to examine the relationship of VWF with pro-inflammatory markers such as interleukin-10.
Conclusion
VWF plays an essential role in the occurrence of VOC, hemolysis, anemia, and inflammation in patients with SCD. The von Willebrand factor plays an important role in determining the clinical severity of sickle cell disease, thus constituting a putative therapeutic target.
Authors’ contributions
Conceptualization: Theresa Nwagha.
Data curation: Theresa Nwagha, Fledgelight Evidence Consult (FEC)
Formal analysis: Fledgelight Evidence Consult (FEC),
Funding acquisition: Theresa Nwagha.
Writing – original draft: Theresa Nwagha, Fledgelight Evidence Consult (FEC), Eyiuche Ezigbo.
Supplemental Appendix
Download MS Word (41.8 KB)Acknowledgements
The authors thank our project research assistant, Ukwuoma Maryjane, for her assistance in data screening and extraction
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Akaba K, Inyama M, Ekwere T, et al. Haemostatic disorders in sickle cell disease subjects in Nigeria: A review of literature. Int Blood Resour Rev. 2018;8:1–7.
- Clancy S. Genetic mutation. Nature Educ. 2008;1(1):187.
- Chen J, Hobbs WE, Le J, et al. The rate of hemolysis in sickle cell disease correlates with the quantity of active von Willebrand factor in the plasma. Blood. 2011;117:3680–3683.
- Kato GJ, McGowan V, Machado RF, et al. Lactate dehydrogenase as a biomarker of hemolysis-associated nitric oxide resistance, priapism, leg ulceration, pulmonary hypertension, and death in patients with sickle cell disease. Blood. 2006;107:2279–2285.
- Akaba K, Nwogoh B, Oshatuyi O. Determination of von Willebrand factor level in patient with sickle cell disease in vaso-occlusive crisis. Res Practice Throm Hem. 2020;4:902–905.
- Ataga KI, Key NS. Hypercoagulability in sickle cell disease: new approaches to an old problem. Hematology. 2007;1:91–96.
- Favaloro JE, Henrey BM, Lippi G. Covid-19 and antiphospholipid antibodies Time for a reality check? Seminar Throm Hem. 2022;48:72–92.
- Zhou Z, Han H, Cruz MA, et al. Haemoglobin blocks von Willebrand factor proteolysis by ADAMTS-13: a mechanism associated with sickle cell disease. Thromb Haemost. 2009;101(6):1070–1077.
- Omer NE, Maria M, Satti H, et al. Plasma level of von Willebrand factor: an indicator of severity in sickle cell disease. Sudan JMS. 2009;4(2). Doi:10.4314/sjms.v4i2.44897.
- Chen J, Fu X, Wang Y, et al. Oxidative modification of von Willebrand factor by neutrophil oxidants inhibits its cleavage by ADAMTS13. Blood. 2010;115:706–712.
- Ahmed SG, Kagu MB, Ibrahim UA. Correlation between ABO blood group and vaso-occlusive crisis among adult patients with sickle cell anaemia in northern Nigeria; 2010. Egyptian J Haematol. 2014;39:227–231.
- Studt JD, Kremer-Hovinga JA, Antoine G, et al. Fatal congenital thrombotic thrombocytopenic purpura with ADAMTS 13: in vitro inhibitor of ADAMTS 13 activity by haemoglobin. Blood. 2005;105:542–544.
- Shamseer L, Moher D, Clarke M, et al. Preferred reporting items for systematic review and meta-analysis protocols (PRISMA-P) 2015 statement. Sys Rev. 2015;2015(4):1.
- Hong QN, Fàbregues S, Bartlett G, et al. The mixed methods appraisal tool (MMAT) version 2018 for information professionals and researchers. Educ Inf. 2018;34(4):285–291.
- Higgins J, Thompson S. Quantifying heterogeneity in a meta-analysis. Stat Med. 2002;21:1539–1558.
- Higgins J, Thompson S, Deeks J, et al. Measuring inconsistency in meta-analyses. Br Med J. 2003;327:557–560.
- Higgins JPT, Thomas J, Chandler J, et al. Cochrane Handbook for Systematic Reviews of Interventions version 6.2 (updated February 2021). Cochrane, 2021.
- Demagny J, Aurélie D, Alain S, et al. ADAMTS13 and von Willebrand factor assessment in steady state and acute vaso-occlusive crisis of sickle cell disease. Res Pract Thromb Haemost. 2021;5:197–203.
- Mohan JS, Lip PL, Blann AD, et al. The angiopoietin/Tie-2 system in proliferative sickle retinopathy: relation to vascular endothelial growth factor, its soluble receptor Flt-1 and von Willebrand factor, and to the effects of laser treatment. Br J Ophthalmol. 2005;89:815–819. doi: 10.1136/bjo.2004.058164.
- Blanna AD, Marwah S, Serjeant G, et al. Platelet activation and endothelial cell dysfunction in sickle cell disease is unrelated to reduced antioxidant capacity. Blood Coagul Fibrin. 2003;14:255–259.
- Dong JF, Moake JL, Nolasco L, et al. ADAMTS-13 rapidly cleaves newly secreted ultralarge von Willebrand factor multimers on the endothelial surface under flowing conditions. Blood. 2002;100(12):4033–4039. doi: 10.1182/blood-2002-05-1401.
- Arya M, Anvari B, Romo GM, et al. Ultralarge multimers of von Willebrand factor form spontaneous high-strength bonds with the platelet glycoprotein Ib-IX complex: studies using optical tweezers. Blood. 2002;99(11):3971–3977. doi: 10.1182/blood-2001-11-0060.
- Mohandas N, Evans E. Sickle erythrocyte adherence to vascular endothelium: morphologic correlates and the requirement for divalent cations and collagen-binding plasma proteins. J Clin Invest. 1985;76(4):1605–1612.
- Wick TM, Moake JL, Udden MM, et al. Unusually large von Willebrand factor multimers increase adhesion of sickle erythrocytes to human endothelial cells under controlled flow. J Clin Invest. 1987;80(3):905–910.
- Fujikawa K, Suzuki H, McMullen B, et al. Purification of human von Willebrand factor-cleaving protease and its identification as a new member of the metalloproteinase family. Blood. 2001;98(6):1662–1666.
- Browne PV, Mosher D, Steinberg MH, et al. Disturbances of plasma and platelet thrombospondin levels in Sickle cell anaemia. Am J Haematol. 1996;51:296–301.
- Richardson SG, Mathews KB, Stuart J, et al. Serial changes in coagulation and viscosity during sickle cell crisis. Br J Haematol. 1979;41:95–103.
- Bolanos-Meade J, Keung YK, Lopez-Arvizu C, et al. Thrombotic thrombocytopenic purpura in a patient with sickle cell crisis. Ann Hematol. 1999;78(12):558–559.
- Shelat SG. Thrombotic thrombocytopenic purpura and sickle cell crisis. Clin Appl Thromb Hemost. 2010;16(2):224–227.
- Walmet PS, Eckman JR, Wick TM. Inflammatory mediators promote strong sickle cell adherence to endothelium under venular flow conditions. Am J Hematol. 2003;73(4):215–224.
- Colombatti R, De Bon E, Bertomoro A, et al. Coagulation activation in children with sickle cell disease is associated with cerebral small vessel vasculopathy. PLoS ONE. 2013;8(10):e78801. doi:10.1371/journal.pone.0078801.
- Sins JWR, Schimmel M, Luken BM, et al. Dynamics of von Willebrand factor reactivity in sickle cell disease during vaso-occlusive crisis and steady state. J Thromb Haemost. 2017;15(7):1392–1402. doi: 10.1111/jth.13728.
- Krishnan S, Siegel J, Pullen G, Jr, et al. Increased von Willebrand factor antigen and high molecular weight multimers in sickle cell disease associated with nocturnal hypoxemia. Thromb Res. 2008;122(4):455–458.
- Inacio P. (2020). Inflammation Markers Identify Sickle Cell Patients at Risk for Worse Outcomes, Study Suggests. https://sicklecellanemianews.com/2020/03/31/inflammation-markers-identify-sickle-cell-patients-at-risk-of-worse-outcomes-study-suggests/.
- Domingos IF, Pereira-Martins DA, Sobreira MJVC, et al. High levels of proinflammatory cytokines IL-6 and IL-8 are associated with a poor clinical outcome in sickle cell anemia. Ann Hematol. 2020;99:947–953. https://doi.org/10.1007/s00277-020-03978-8.